
Abstract
The synthesis of a chiral periodic mesoporous organosilica (chiral PMO) SBA-15 type, using a bis-silylated Binol precursor, has been studied through two different heterogenization routes. On the one hand, the immobilization of a (R)-(+)-1,1′-bi-2-naphthol (Binol) derivative was accomplished by following a standard multistep synthesis methodology. On the other hand, a new route consisting of an easy one-step synthesis was developed achieving a simultaneous formation of mesoporous structure and Binol chiral ligand immobilization in the walls. The addition of KCl “salting out” electrolytes favored the micellization, obtaining the well-ordered chiral PMO materials. The thioanisole asymmetric oxidation reaction was used to validate the enantio-catalysts activity. The materials synthesized by the multistep method reached yield and enantiomeric excess of 41 and 15 %, respectively, while the synthesized one-pot chiral PMO materials achieved up to 58 and 42 %, respectively. This difference could be attributed to a more homogenous distribution of the chiral moiety as well as the simultaneous micellization and formation of the siliceous mesostructure, in the one-pot procedure. This promotes higher reagents accessibility to the active center, and therefore an enhancement of the chiral induction. Thereby, materials with Binol ligand incorporated into the three-dimensional silica framework were successfully accomplished.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the discovery of asymmetric catalysis by Knowles and Kagan [1] in the late early 1970s, a number of articles emerged in this specific field, as asymmetric catalysis is one of the most general and interesting strategies for the synthesis of modern pharmaceuticals and agrochemicals. In this sense, heterogeneous catalysis will also play a key role in creating a sustainable society going forward due to the easy purification of the product and recycling of the chiral catalysts [2]. However, the economy of the process would not only require the reduction of waste production but also require similar activities and enantioselectivities to the homogeneous catalysts. In the last years, the heterogeneous catalysts have contributed to the development of fine chemicals alternative production processes. In this context, the zeolites have acquired great relevance as it is possible to tune their catalytic properties, such as hydrophobicity and pore size (shape selectivity) [3]. The main drawback of these materials emerges when using bulky reactants, larger than their pore size. Thus, the discovery of mesoporous materials M41S family has brought a revolution in the heterogeneous catalysis, as its larger pore size allows to work with cumbersome molecules present for instance in the petrol crude and also with bulky compounds in the fine chemical sector [4–6]. The easier procedure to immobilize some organic moieties consists of either the impregnation or ionic interexchange; however, the interaction between the solid support and organometallic complex is too weak, leading to a great amount of leaching [7]. However, the most part of the investigations are devoted to the immobilization of organic complexes by grafting (covalent anchoring) to the silanols groups of the silica support and by direct synthesis or co-condensation of silicon and organosilicon moieties [8, 9]. Grafting via heterogenization requires the anchoring of the catalytic complex through a position which does not alter its catalytic activity, resulting often in the need to make modifications of the solid support by a tethering or bridge groups [10, 11]. These compounds are organic molecules that separate the active site of the catalyst from the support solid wall, reducing the possible interactions. Corma et al. [12, 13] have employed this methodology to heterogenize different homogeneous catalysts, such as the chiral Salen catalyst. Direct synthesis strategy is a better alternative when higher loadings are needed. Thus, different alkoxysilanes as organic precursors together with silica precursor, mainly tetraethylorthosilicate (TEOS), are condensed in the same step, allowing larger amounts of organic incorporation onto the silica walls. Another interesting challenge has been the incorporation of these catalytic organic moieties into the porous structure of the organic–inorganic silica-based material, named periodic mesoporous organosilica (PMO), through the use of bis-organosilane precursors. In 1999, different research groups discovered a new class of siliceous mesostructured materials, which contain a homogeneous distribution of organic and inorganic species inside their tridimensional structure, also a high structural ordering and defined pore diameter size in the interval from 20 to 40 Ǻ [14–16]. The main difference related to the previously mentioned heterogenization procedures lies in the use of bis-organosilanes, i.e., organic moieties with two alkoxysilanes in the chain. These types of compounds allow the incorporation of the organic ligand in the tridimensional structure of the silica matrix, whereas in the previous procedures the organic moiety is located on the external wall of the inorganic silica. Accordingly, an adequate organic ligand choice allows obtaining a great variety of different materials with specific textural and catalytic properties. That is why PMOs are claimed as novel and interesting materials with applications as selective adsorbents, chromatographic separations, and powerful catalysts [17, 18]. In the case of the asymmetric catalysis, the direct anchoring of the chiral organic ligand into the tridimensional structure of the silica phase through the use of the bis-silylated chiral precursor lead to chiral PMOs. These materials incorporate the chiral organic moiety as constituent of the material structure, inducing the asymmetric feature, and consequently, becoming and behaving as chiral solid catalysts. This new heterogenization procedure incorporates a higher amount of organic precursor in the solid support [19–33].
A versatile ligand that has found widespread use in asymmetric catalysis is (R)-(+)-1,1′-bi-2-naphthol or (R)-Binol [34, 35]. Both Binol enantiomers are commercially available and have been extensively used as chiral ligand in catalysis, especially in combination with titanium salts. The Binol/Ti system catalyzes a number of asymmetric reactions including allyl and Mukaiyama–aldol additions to aldehydes [36], hetero Diels–alder reactions, ene-reaction, reduction of ketones and aldehydes, and oxidation of sulfides [37, 38]. Chiral Binol was also immobilized on a number of supports such as polymers, dendrimers, siliceous supports, and ionic liquid systems [39–41]. Every heterogeneous methodology deals with (a) the strategy to support chiral Binol, (b) the catalytic activity of these supported catalysts, (c) minimizing undesired catalytic activity, and (d) regeneration of the expensive chiral catalyst [42]. The integration of (R)-(+)-Binol in the mesoporous framework may further reveal the influence of the rigid porous wall on the enantioselectivity of the supported chiral catalyst, which is very important to design and synthesize efficient solid catalyst for asymmetric catalysis [31]. In 2004, García et al. [12] described the incorporation of (R)-(+)-Binol into the framework of PMOs materials. However, in most of the heterogenization attempts, the OH groups of the (R)-(+)-Binol were chosen as the sites to be derivatized, which occupied the coordination position and hindered the coordination of Binol to metal. With the aim of modifying the framework composition of the chiral PMOs and immobilizing the Binol moieties from the position different than the hydroxyl groups, Yang et al., described the Binol modification through the 6,6′ positions via multistep reactions using trimethoxyvinylsilane as linker group. After coordination to Ti(OiPr)4, these chiral PMOs catalyzed the asymmetric addition of diethylzinc to benzaldehyde with high yields and moderate enantioselectivity [22].
In the study described herein, we report on the synthesis of a PMO and its simultaneous functionalization with a Binol derivative, in the same reaction acidic medium. The synthesis of the material proceeds through the transamidation reaction of the Binol derivative with the organosilane (N-methylaminopropyl)-trimethoxysilane to form the corresponding chiral bis-organosilane, and simultaneously its hydrolysis, together with the TEOS, and the condensation of both around the micelles of structure-directing surfactant. The result is the formation of a PMO material functionalized with chiral Binol derivative incorporated in its structure in only one step. In addition, we present the comparative result with a chiral mesoporous material, prepared following a multistep synthesis methodology to obtain the chiral Binol ligands also incorporated into the silica framework. Finally, the study is aimed toward the comparison of the results of both materials as catalysts in asymmetric sulfoxidation reactions, and therefore the different role that the structure and steric constrictions of both organically modified materials may exert on the enantioselectivity achieved.
Experimental
Chemicals
Pluronic P123 (PEO20-PPO70-PEO20; Aldrich), tetraethylorthosilicate (TEOS, Aldrich), potassium chloride (KCl, 99 %, Aldrich), hydrochloric acid (HCl, 35 %, Scharlab), n-butyl stannonic acid (99 %, Aldrich), cumyl hydroperoxide (CHP, Aldrich), methyl phenyl sulfide (99 %, Aldrich), n-methyl aminopropyl trimethoxysilane (GELEST), dimethyl-2,2′-dihydroxy-1,1′-binaphthalene-3,3′-dicarboxylate (98 %, Aldrich), and ethanol (Scharlab) were used as received without further purification. Dichloromethane was distilled from P2O5 prior its use as solvent for catalytic test.
Chiral PMO multistep methodology
First step: synthesis of the chiral bis-silane
Every synthesis steps were carried out in oven-dried glassware under nitrogen atmosphere. Dimethyl-2,2′-dihydroxy-1,1′-binaphthalene-3,3′-dicarboxylate (0.59 g) was reacted with two equivalents of N-methyl aminopropyl trimethoxysilane (0.61 g) in toluene under reflux conditions. The reaction was catalyzed by n-butyl stannonic acid (0.1 eq.) [27] and using a Dean-Stark apparatus for solvents lighter than water for the removal of methanol by azeotropic distillation, to carry out the transamidation reaction shown in Scheme 1. The reaction was monitored by TLC and continued until completion. The double silane-functionalized Binol was obtained as an orange solid product and used after filtration for catalyst elimination.

Synthesis of bis-silylated chiral Binol derivative
Second step: chiral PMOs synthesis
In a regular synthesis, pluronic 123, hydrochloric acid, and water were magnetically stirred in a beaker until complete dissolution. After complete dissolution, the mixture was warmed up to 40 °C. To the transparent resultant solution, TEOS, KCl, and the bis-silylated Binol precursor were added separately by dropping. The resultant gel was then stirred for 20 h and afterward transferred to an autoclave to be aged. The aging step of the gel was carried out under static conditions at 110 °C and autogenous pressure for 24 h. The resultant materials were recovered by filtration and air-dried. As-made material was treated with an ethanolic solution of hydrochloric acid for the surfactant removal.
Chiral PMO one-step methodology
In a typical synthesis, pluronic P123 and KCl were dissolved in hydrochloric acid in a round-bottom flask at room temperature. After complete dissolution, the mixture was warmed up to 40 °C and TEOS were added in a single step. After 1 h, (N-methyl-3 aminopropyl) trimethoxysilane and the Binol precursor, dimethyl-2,2′-dihydroxy-1,1′-binaphthalene-3,3′-dicarboxylate, were added to obtain the bis-silylated Binol derivative through in situ transamidation reaction. Different materials were prepared to study the synthesis conditions in the materials ordering in terms of: KCl concentration, HCl concentration, and the molar range TEOS to Binol tartramide. Then, the solution was vigorously stirred for 20 h at 40 °C and hydrothermally aged at 110 °C for another 24 h. The product was then recovered by filtration and air-dried. The surfactant removal consisted on an extraction step with ethanol. Typically, 1 g of as-made material was treated with 100 mL of ethanol under reflux overnight. The solids were then recovered by filtration while still warm and thoroughly washed with fresh ethanol before air-dried.
Characterization techniques
The so-prepared materials were characterized by means of different analytical techniques. Thus, nitrogen adsorption–desorption isotherms were collected at 77 K using a Micromeritics TriStar 3000 unit. Previously, the samples were outgassed at 200 °C for 4 h under nitrogen flow. The surface area measurements were performed according to the BET method from nitrogen adsorption points in the range P/P 0 = 0.05–0.2. Pore size distribution was determined applying the Barrett–Joyner–Halenda model (BJH) applied to the adsorption branch of the isotherm, assuming cylindrical pore geometry. For each sample, the average pore size was estimated as the diameter corresponding to the maximum of the pore size distribution curve. Total pore volume was taken at a relative pressure P/P 0 of 0.985.
X-ray powder diffraction patterns were collected on a Philips X’pert diffractometer equipped with an accessory for low angle measurements. XRD analyses were recorded using the Cu Kα line in the 2θ range from 0.5° to 10° with a step size of 0.02° and a counting time of 10 s.
FTIR analyses were performed, using the KBr buffer technique, on a Mattson Infinity series apparatus in the wavelength range from 4000 to 400 cm−1 with a step size of 2 cm−1 and collecting 64 scans for each analysis. The samples were also placed in a catalytic chamber HVC-DPR (Harrick Scientific Company) where they were heated at different temperatures under vacuum (<10–4 mbar) with a 1.5 °C/min heating rate.
Solid-state 13C and 29Si MAS NMR experiments were performed on a Varian Infinity 400 MHz spectrometer fitted with a 9.4 T magnet. These nuclei resonate at 100.53 and 79.41 MHz, respectively. An H/X 7.5 mm MAS probe and ZrO2 rotors spinning at 6 kHz were used. On CP experiments, the cross-polarization time was determined to guarantee the total proton polarization verifying the Hartmann–Hann condition. For 13C acquisition, π/2 pulse, number of scans, repetition delay, and contact time were 4.25 μs, 2000 scans, 3 s, and 1 ms, respectively. The 29Si CP experiments were performed for 3000 scans, π/2 pulse of 3.5 μs and 15 s of repetition time, while the contact period was 10 ms as cross-polarization depends upon heteronuclear dipolar interaction, the greater distance the larger cross-polarization time. 13C and 29Si chemical shifts were externally referenced to adamantane and tetramethylsilane, respectively.
Catalytic test
Reaction tests for the asymmetric oxidation of sulfides were carried out under nitrogen atmosphere using standard Schlenk techniques. The PMO used as solid chiral ligand was previously outgassed and dried in a Kugelrhor apparatus at 130 °C under vacuum (1 Torr) overnight before being employed in the catalytic tests. Inert conditions were ensured by passing a continuous nitrogen stream through the reactor which was maintained during the overall operation. Freshly distilled dichloromethane (50 mL) was then transferred into the reactor via syringe and the mixture magnetically stirred. Titanium isopropoxide (0.056 mmol) and 2-propanol were added drop by drop to the resultant suspension. The mixture was then aged for 2 h to promote the contact between the titanium species and the chiral ligand sites present in the PMO material. After the aging step both cumyl hydroperoxide (CHP, 0.56 mmol) and methyl phenyl sulfide (0.56 mmol) were added separately by dropping onto the catalytic suspension. Sample aliquots were collected to assess the evolution of the reaction. The mixture was allowed to react during 24 h.
Results and discussion
One-step chiral PMOs synthesis route
The periodic mesostructured organosilicas with chiral moieties incorporated into the framework were obtained following a recent and an easy one-pot methodology previously reported [33]. To optimize the experimental conditions, the effect of the acidic media on the structural order and chemical composition of the resultant materials were investigated due to the instability of the amide bond under strong acidic media. The commonly strong acidic media used for the SBA-15 material synthesis (1.9 M HCl) led to no chiral bis-organosilane incorporation, as the C–N bonds were broken during the procedure, as IR spectrum (not shown) demonstrated. In addition, experiments accomplished under almost neutral pH showed that the absence of acidity was unable to hydrolyze the silicon species, resulting in no solid precipitation during the material preparation. Nevertheless, working under weak acidic media (0.5 M HCl) the material exhibits one sharp diffraction pattern in its XRD pattern at 2θ = 0.8°–0.9°, corresponding to the d 100 basal plane of SBA-15 type material. IR spectrum (not shown) also confirms the Binol integration in the silica frame, as the signals corresponding to the aryl and carbonyl groups at around 1200–1600 and 1640 cm−1, respectively [43], are clearly visualized.
Once the sensibility of the one-pot methodology to the pH of the synthetic medium was studied, the effect of the inorganic salt addition in the chiral PMOs was investigated. Currently, although great effort has been spent on the preparation of PMOs SBA-15 type, in most cases these products were either poorly ordered or the synthesis was necessarily carried out under somewhat high critical conditions of acidity or surfactant concentration [44]. Following our previous studies [33, 45], an easy synthesis pathway to prepare ordered chiral PMOs SBA-15 type with bulky organosilane groups in the walls using “salt-assisted” self-assembly concept is studied in this article. Materials with different KCl/Si ratios have been explored to prove the importance of the inorganic salt addition when treating bulky bis-organosilanes, keeping the ratio TEOS:BINOL = 1:0.2 constant for all the materials prepared. Figure 1 shows the N2 adsorption–desorption isotherms, and XRD patters of the samples synthesized with and without salt.

Nitrogen adsorption–desorption isotherms and pore size distribution recorded for chiral one-pot PMOs synthesized under different KCl/Si ratios
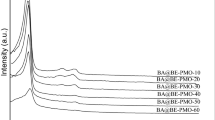
Well-ordered chiral PMOs can be easily synthesized with the aid of “salting effect” over the wide range of KCl/Si molar ratio from 1 to 6, clearly indicating the important effect of the inorganic salts on the assembly process of ordered chiral PMO materials, favoring the micellization [44, 46]. Between these ratios, all samples exhibit isotherms resembling type IV with a nearly parallel H1 hysteresis loop. This strongly suggests the presence of uniform cylindrical pores in these materials, in accordance with the two-dimensional hexagonal pore structures revealed by the XRD data (Fig. 2). The ratio KCl/Si = 3 provides a material with better textural and structural properties, according to XRD patterns and N2 adsorption–desorption isotherms results. In contrast, working with no “salt-assisted” conditions (KCl/Si = 0) leads to hardly detectable reflection peaks in XRD patterns, and also poor physic-chemical properties values due to the lack of order in the PMO structure, as displayed in Table 1.

XRD patterns recorded for chiral one-pot PMOs synthesized under KCl/Si ratios = 0–6
Upon fixing the optimal pH and KCl/Si ratio, another important parameter to be studied in the preparation of Binol PMOs SBA-15 type materials is the maximum organic loading that the chiral PMOs structure can accommodate without significant losing of the ordering degree. For this purpose, several loadings of Binol bis-organosilane precursor were used, ranging from 10 to 40 % of organosilane to TEOS ratio on a molar basis (Table 1). N2 adsorption–desorption tests revealed the dramatic influence of the organosilicon amount on the final textural properties of the PMOs materials. Therefore, increasing the organic loading promotes a large decrease on the surface area, pore size, and pore volume. Whereas low organic loading leads to well-structured materials, increasing the amount of Binol precursor causes not only a great decrease on all the textural properties, but also the isotherm shape is also shifted. Thus, from sample containing 10 to 40 % mol of Binol functionalities, the hysteresis loops become wider, indicating the partial framework destruction, similar tendency to the tartrate PMOs materials previously synthesized [45].
The increase of the organic loading shows some consequences on the quality of the structure ordering as displayed in TEM images (Fig. 3). TEM images of the material with Binol incorporation up to 20 % (IN-SITU BINOL 10 and 20, Table 1) clearly display the two-dimensional hexagonal arrangement of the mesopores throughout the sample. A honeycomb structure as well as parallel fringe pore wall is noticed in both directions, parallel and perpendicular to the pore axis. However, using 40 mol% of BINOL leads to worm-like pore structure materials. An excessive amount of bulky Binol precursor causes distortion in the structure-directing agent micelles, and therefore, the mesophase ordering is lower than that achieved by a PMO material with smaller bis-organosilanes loading.

TEM images recorded for chiral one-pot PMOs synthesized with Binol/TEOS ratio = 10–40
To further clarify the composition of the one-pot PMO materials prepared, 29Si MAS NMR and 13C CP-MAS NMR were performed (Fig. 4). 29Si MAS NMR spectrum of IN-SITU BINOL-20 shows both the T and the Q silicon sites. The main signals at −112.3 ppm, the peak at −103.5 ppm, and the shoulder peak at −90.3 ppm correspond to Q4, Q3, and Q2, respectively. The broad peak centered at −65.3 ppm could be ascribed to the mixture of T3 and T2 organosilicon species. A large amount of cross-linked T3 and T2 surface species, compared to T1 are formed, indicating that the level of condensation reached for these materials is reasonably high. The absence of T0 site confirms that the chiral moiety is actually integrated both ends in the pore walls, instead of dangling in the pore as a pendant group. As the 29Si NMR spectrum was obtained by a direct one pulse experiment, the quantification of the species associated with the respective intensities can be obtained from the deconvolution of the spectrum, as shown in Fig. 4.

29Si MAS NMR (left) and 13C CP-MAS NMR (right) solid-state spectra of the one-pot IN-SITU BINOL 20 sample (below the Binol precursor 13C liquid NMR spectrum)
The peak area ratio of (T/2)/((T/2) + Q) = 15.3 %, which agrees well with the molar chiral precursor concentration in the initial gel (20 %). The weak acidic synthesis conditions, 0.5 M HCl, seem to be enough to almost incorporate stoichiometrically the chiral Binol precursor in the silica mesoporous network for samples prepared with organic concentrations below 20 mol%. In addition, the concentration of the chiral organic moiety in the silica framework was calculated as mmol Binol/g material, as displayed in Table 1. Increasing the Binol moieties loading induces a negative influence on the ordering of the final materials without enhancing the incorporation degree of the chiral functionality. The results point out an optimum ratio of TEOS to chiral precursor to be used to achieve high incorporation degrees of the chiral ligand in well-structured materials. This optimal molar ratio is reached for 80:20 TEOS/bis-silylated Binol molar ratio, probably as consequence of the voluminous chiral precursor employed. In previous investigation, chiral tartrate PMOs materials with a ratio Si/tartrate as high as 50:50 have been synthesized, which suggest the strong dependence of the chiral precursor volume in the textural properties decline [33].
The identification of these organic species incorporated into the framework of mesostructured materials can be inferred from the 13C CP-MAS NMR spectra. Figure 4 depicts the 13C CP-MAS NMR spectrum recorded from IN-SITU BINOL-20. This figure shows the signals in the range of δ = 160–110 ppm ascribed to aryl carbon atoms of Binol precursor and the lower shielding chemical shift at about 178 ppm corresponds to quaternary carbons at the carbonyl groups, C=O, influenced by the nitrogen atom from the synthesized amide groups. The signals around 10–25 ppm are assigned to Si–CH2, Si–CH2–CH2, and Si–CH2–CH2–CH2. The spectrum also shows one peak centered at 50 ppm that could be related to the presence of N–CH3, which indicates the presence of the nitrogen atom in the material according with the Binol homogeneous 13C NMR spectra. Nevertheless, all this data confirm the presence of the immobilized Binol organic precursor in the inorganic material framework by the one-pot methodology. In the one-pot procedure, once the chiral Binol precursor is mixed with the (N-methyl-3-aminopropyl) trimethoxysilane under weak acidic conditions (0.5 M HCl) needed for synthesizing a SBA-15 like framework material, a transamidation reaction takes place, leading to the bis-silylated chiral precursor. Besides, the presence of TEOS and the block copolymer P123 provides the mesoscopic ordering to the final material. Therefore, during the in situ transamidation reaction, the hydrolysis of the chiral bis-silane and the silica precursors and the condensation of these latter species around micelles formed by the surfactant leads to a chiral PMO material following this easy one-pot protocol.
Multistep co-condensation chiral Binol PMO methodology
To compare the textural and structural properties showed in one-pot synthesized materials, chiral PMOs prepared under a multistep procedure have been achieved. The mesostructured organosilicas with chiral Binol ligands incorporated into the framework were obtained following a direct co-condensation procedure through the surfactant-assembling pathway, distributed in different steps. First, the synthesis of modified Binol to be incorporated into the PMO structure is required. Thus, (N-methyl-3-aminopropyl)-trimethoxysilane was reacted with the chiral derivative Binol as shown in Scheme 1 (“Experimental” section). Further tedious quenching and purification gives the bis-silylated Binol chiral ligand as shown in Fig. 5, where we have compared the 13C NMR spectrum of the starting materials together with the Binol bis-silane obtained. Subsequently, in another different step, the previously synthesized Binol precursor was used in combination with TEOS to prepare chirally modified SBA-15 PMO type materials, using pluronic P123 as structure-directing agent, the ratio of chiral moiety related to TEOS being 20:80, i.e., the optimum for one-step synthesized Binol PMOs. Figure 6 (left) shows the N2 adsorption–desorption isotherm, as well as pore size distribution for the prepared sample. The curve displays type IV isotherm, according to the IUPAC classification typical for mesoporous solids. For comparative purpose with materials prepared under one-pot methodology, sample IN-SITU BINOL 20 has been included in Table 2. The textural properties show that one-pot methodology leads to a greater control of surface properties and presents a larger incorporation of chiral organic species, compared with multistep method, as shown in Table 2. The periodic structure of the template-free chiral PMO was determined by powder X-ray diffraction (Fig. 6, right). The presence of the characteristic diffraction at 2θ = 0.8°–0.9°, corresponding to the d 100 basal plane of SBA-15 type material, confirms the formation of the mesoporous structure for this material synthesized under the multistep methodology.

13C NMR spectrum of the starting material and product resulting of the chiral bis-silane synthesis reaction (a) N-methyl aminopropyltrimethoxysilane (starting material); (b) dimethyl-2,2′-dihidroxy-1,1′-binaphtalene-3,3′-dicarboxilate (starting material); and (c) CP-MAS 13C NMR bis-silylated Binol derivative

Nitrogen adsorption–desorption isotherms and pore size distribution (left). XRD patterns (right) recorded for chiral PMO multistep sample
The solid-state 13C CP-MAS spectrum of the MULTI-STEP BINOL-20 material shows the presence of the organic moieties in the solid (Fig. 7). The lower shielding chemical shift at about 180 ppm corresponds to quaternary carbons at the carbonyl groups, C=O, from the synthesized amide during the chiral bis-silane synthesis. The set of chemical shifts at 156, 133, 129, and 118 ppm may be assigned to the binaphthyl groups. The aromatic carbon species attached to the OH groups are found at 155 ppm. The signal centered at 32 and 51 ppm can be assigned to NCH3 and NCH2, respectively, which indicates the presence of the nitrogen atom in the material. Also, the two sharp peaks centered at 10 and 19 ppm were assigned to Si–CH2 and Si–CH2–CH2, respectively. The spectrum displays different signals that have been assigned to the different functional groups of the silica-supported Binol chiral species, all of them similar to the ones assigned for the material prepared following the one-step methodology. Both T and Q silicon sites were clearly observed in the 29Si MAS NMR spectra of MULTI-STEP BINOL-20. The signals at −110.6, −103.1, and −92.1 ppm may be attributed to the Si species Q4 (≡SiO)4Si, Q3 (≡SiO)3–Si(OR′), and Q2 (≡SiO)2Si(OR′)2, while the signals at −70.1 and −64.5 ppm can be ascribed to the organosilicon species T3 (R-Si(OSi≡)3) and T2 (R-Si(OSi≡)2), respectively. Moreover, the absence of the T0 sites in the 29Si MAS NMR spectra of MULTI-STEP BINOL-20 indicates the absence of pendant groups. As it is known, the amount of T groups is directly related to the presence of organic functionalities. Therefore, the functionalization degree can be measured as the ratio between the calculated area for T groups referred to the area below Q and T groups. The concentration of Binol groups for MULTI-STEP BINOL-20 sample is 0.6 mmol Binol/g, calculated by 29Si NMR. The above characterization confirms the successful synthesis of chiral PMOs with Binol moieties integrated in the framework, following the multistep methodology.

29Si MAS NMR (up) and 13C CP-MAS NMR (down) solid-state spectra of the multistep chiral PMO BINOL-20
Catalytic test
To determine the enantioselective properties of the materials, and the influence of the synthesis methodology, multistep and one-pot protocol, the thioanisole sulfoxidation reaction was chosen as catalytic test, in the presence of cumene hydroperoxide (CHP) as oxidizing agent as shown in Scheme 2. In this reaction, products are obtained as sulfoxide R and S enantiomers, and the by-product methylsulfone, as the result of complete oxidation of thioanisole. The methodology proposed by Katsuki [47] was selected as starting point to evaluate the catalytic systems. Organosulfur compounds, such as sulfoxides and sulfones, are useful synthetic reagents in organic chemistry. In particular, sulfoxides are important intermediates in the synthesis of natural products and biologically significant molecules [48], and they have also been employed as ligands in asymmetric catalysis [49]. An application is the synthesis of the enantiomerically pure S-enantiomer of omeprazole. This enantiomer is called esomeprazole, highly potent gastric acid secretion inhibitor.

Catalytic test: asymmetric oxidation of methyl phenyl sulfide (thioanisole)
Based on the optimized experimental conditions obtained in previous investigations [33, 45], the synthesized materials MULTI-STEP BINOL-20 and IN-SITU BINOL-20 were tested in the thioanisole sulfoxidation reaction. The chiral ligand concentration was constant for both catalysts in each reaction. The presence of more than 1 eq. of alcohol with respect to sulfide was essential for the oxidation, and it was found that the alcohol was necessary not only to produce an effective catalyst for a highly enantioselective oxidation but also to maintain the catalytic activity of the chiral catalyst for longer period of time. Table 3 summarizes the yield and enantiomeric excess (ee) results of the fresh catalyst synthesized by means of the one single step “in situ” methodology (58 and 42 %, respectively, obtaining the R configuration as the most abundant enantiomer). As well as the values were obtained following the multistep methodology (41 and 15 %, respectively). The results show that the sulfone yield is less than 10 % for each reaction. It is remarkable the higher ee of the one-pot material, which is induced to the sulfide groups. The more homogeneous distribution of the different compounds used to synthesize the Binol chiral precursor, together with the silica precursor in the same reaction media endorses an enhancement of the chiral induction over the reagents, like alkyl phenyl sulfides, as a higher accessibility to the active centers seems to be promoted. The local environment of the catalytic site is important in directing the enantioselectivity of the reactions. In this sense, the multistep methodology might conduce to chiral environments with more interactions with the silanols groups of the silica surface, inducing weak metal–chiral ligand interaction. These hypotheses are based on the explanation that several authors offer about the Ti-tartrate complex structure, i.e., the Sharpless and Kagan catalyst, and it could be translate to the Ti-Binol complex used in the reactions. Ti-tartrate catalyst affords consistent enantioselectivity for widely different substrates by virtue of a combination of stereoelectronic, geometrical orientation, and steric factors. The origin of enantioselectivity was reported to be based on the construction of the more rigid chiral backbone the better. This provides different steric proposal, first, the bulky oxidant (cumyl hydroperoxide) is forced to adopt a single orientation when chiral backbone is more rigid. Second, the corresponding sulfide is thereby restricted to reaction at a single coordination site on the metal center, which results in a more efficient catalytic turnover and enantioselectivity.
Taking into account textural properties, the catalytic difference could be attributed not only to the homogeneous dispersion of the chiral moiety but also to the lower structural order and BET surface area of the multistep materials, comparing materials with the same chiral organic precursor in the synthesis media. However, to avoid the textural properties influence we have compared two materials: MULTI-STEP BINOL-20 and IN-SITU BINOL-30. Both materials present similar textural properties, in terms of BET surface area, pore volume, and pore diameter, and moreover the chiral ligand concentration was constant for both catalysts in the reactions. The results are summarized in Table 3. As shown, the higher yield and ee were achieved for the IN-SITU BINOL-30. As consequence, results evidenced the conclusion outlined in the manuscript. The homogeneous chiral moieties dispersion clearly influences and enhances the catalytic results. Furthermore, the higher structural order of the one-step material is another parameter to account for its better catalytic performance in comparison to multistep method. Both parameters provide more rigid chiral backbone, which is the origin of the enantioselectivity.
In addition to the fresh catalysts results, re-use tests were performed over both catalytic systems. The catalysts were recovered after reaction, and washed with different polar and non-polar solvents, being tested after drying overnight. Results clearly indicated that the materials synthesized through the one-pot methodology exhibited better results, in terms of both, yield and ee, than the materials synthesized under the multistep methodology.
Conclusions
In summary, we have successfully incorporated chiral Binol units on a SBA-15 type material by two synthesis routes. The one-step method provides the synthesis of chiral PMO materials with a greater control of surface properties and it also presents larger incorporation of chiral organic species (up to 1.21 mmol Binol/g material compared to 0.6 mmol Binol/g material for the multistep method). Likewise, FTIR and NMR solid-state techniques confirmed the presence of the chiral Binol precursor in the SBA-15 type one-pot material framework following the two methodologies. In addition, well-ordered one-pot PMO may easily be synthesized throughout the “salting effect” of the KCl salt. This effect clearly indicates the important influence of the salt concentration on the assembly process of ordered PMO materials, which promotes the micellization process. The thioanisole asymmetric oxidation reaction has been used to validate the heterogeneously synthesized catalyst activity. Chiral PMOs materials, synthesized using one-step methodology, displayed the greatest yield and ee in those reactions. These materials achieved ee and yield of 42 and 58 %, respectively, while materials synthesized by the multistep methodology achieved 15 and 41 %, respectively. This difference is attributed to the way in which the chiral active site is incorporated into the material in each methodology. In the one-pot methodology, the same reaction media for the simultaneous Binol chiral precursor and silica syntheses provides a more homogeneous distribution of the chiral moiety. This homogeneity promotes higher reagents accessibility to the active center, causing an enhancement of the chiral induction. The significantly good catalytic results obtained in the one-pot methodology compared with the multistep one lead to suggest that organic Binol groups were mainly placed in the accessible porous wall, instead than in the occluded internal silica framework, which would negatively contribute to the induced enantioselectivity over the final sulfoxides. This approach affords a new synthesis route for incorporating not only Binol moieties but also other homogeneous chiral auxiliaries on the silica three-dimensional mesostructured support by means of an easy one-step route.
References
Kagan HB, Dang T-P (1972) J Am Chem Soc 94:6429
Bein T (1999) Curr Opin Solid State Mater Sci 4:85
Corma A (1997) Chem Rev 97:2373
García RA, van Grieken R, Iglesias J, Morales V, Gordillo D (2008) Chem Mater 20:2964
Prasetyanto EA, Khan NH, Seo H-U, Park S-E (2010) Top Catal 53:1381
Prasetyanto EA, Jeong S-M, Park S-E (2010) Top Catal 53:192
De Vos DE, Dams M, Sels BF, Jacobs PA (2002) Chem Rev 102:3615
Martín-Aranda R, Cèjka J (2010) Top Catal 53:414
Anwander R (2001) Chem Mater 13:4419
Thomas JM, Maschmeyer T, Jonhson BFG, Shepard DS (1999) J Mol Catal A Chem 141:39
Song CE, Lee SG (2001) Chem Rev 102:3495
García H, Corma A, Baleizáo-Gigante B, Das D, Alvaro M (2003) Chem Commun 15:1860
Corma A, Fuentes A, Iglesias M, Morales E, Sánchez F (2006) J Mol Catal A 109:246
Inagaki S, Guan S, Fukushima Y, Ohsuma T, Terasaki O (1999) J Am Chem Soc 121:9611
Melde BT, Holland CF, Blandford A (1999) Chem Mater 11:3302
Yoshiina-Ishii C, Asefa T, Coombs N, Maclachlan MJ, Ozin GA (1999) Chem Commun 24:2539
Inagaki S, Guan S, Ohsuna T, Terasaki O (2002) Nature 416:304
Xia H-S, Zhou C-H, Tong DS, Lin CX (2010) J Porous Mater 17:225
Zhu G, Zhong H, Yang Q, Li C (2008) Microporous Mesoporous Mater 116:36
Inagaki S, Guan S, Yang Q, Kapoor MP, Shimada T (2008) Chem Commun 14:202
Xiao L, Peiyuan W, Yang, Yan Y (2010) Chem-Asian J 5:1232
Wang P, Yang J, Liu J, Zhang L, Yang Q (2009) Microporous Mesoporous Mater 117:91
Mizoshita N, Tani T, Inagaki S (2011) Chem Soc Rev 40:789
Ide A, Voss R, Scholz G, Ozin GA, Antonietti M, Thomas A (2007) Chem Mater 19:2649
Meng X, Yokoi T, Lu D, Tatsumi T (2007) Angew Chem Int Ed 46:7796
Polarz S, Kuschel A (2006) Adv Mater 18:1206
Morell J, Chatterjee S, Klar PJ, Mauder D, Shenderovich I, Hoffmann F, Fröba M (2008) Chem Eur J 14:5935
MacQuarrie S, Thompson MP, Blanc A, Mosey NJ, Lemieux RP, Cathleen CM (2008) J Am Chem Soc 130:14099
Wang TY, Shi JY, Ma BC, Wang W (2010) J Mater Chem 20:6026
Shi JY, Wang CA, Li ZJ, Wang Q, Zhang Y, Wang W (2011) Chem Eur J 17:6206
Liu X, Wang P, Zhang L, Yang J, Li C, Yang Q (2010) Chem Eur J 16:12721
Kuschel A, Polarz S (2010) J Am Chem Soc 132:6558
García RA, van Grieken R, Iglesias J, Morales V, Villajos N (2010) J Catal 274:221
Brunel JM (2005) Chem Rev 105:857
Chen Y, Yekta S, Yudin AK (2003) Chem Rev 103:3155
Heumamm LV, Keck GE (2007) Org Lett 9:4275
Pescitelli G, Bari L, Salvadori P (2006) J Organomet Chem 10:2311
Sahoo S, Kumar P, Lefebvre F, Halligudi SB (2009) J Catal 262:111
Yuan X-Y, Li H-Y, Hodge P, Kilner M, Tastard CY, Zhang Z-P (2006) Tetrahedron 17:240
Pathak K, Ahmad I, Abdi SHR, Kureshy RI, Khan NH, Jasra RV (2006) J Mol Catal A 244:110
Hesemann P, Moreau J (2003) CR Chim 6:199
Pathak K, Bhatt AP, Abdi S, Kuershy RI, Khan NH, Ahmad I, Jasra RV (2006) Tetrahedron 17:1506
Wang P, Liu X, Yang J, Yang Y, Zhang L, Yang Q, Li C (2009) J Mater Chem 19:8009
Zhai S-R, Kim I, Ha C-S (2008) J Solid State Chem 181:67
García R, Morales V, Garcés T (2012) J Mater Chem 22:2607
Xu S, Pu H, Wang H, Han C, Dongquan D, Zhang Y, Luo Y (2012) J Phys Chem Solids 73:1252
Katsuki T (2001) Asymmetric oxidation reactions: practical approach in chemistry. Oxford University Press, New York
Kagan HB (2008) Transition metals for organic synthesis: building blocks and fine chemicals, chap 2.14. Wiley-VCH Verlag GmbH & Co, Weinheim
Shi H, Yu C, He J (2010) J Phys Chem 114:17189
Acknowledgements
The financial support of the Spanish government (CTQ2008-05909/PPQ and CTQ2011-22707) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Morales, V., Villajos, J.A. & García, R.A. Simultaneous synthesis of modified Binol-periodic mesoporous organosilica SBA-15 type material. Application as catalysts in asymmetric sulfoxidation reactions. J Mater Sci 48, 5990–6000 (2013). https://doi.org/10.1007/s10853-013-7395-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7395-5