
Abstract
Drying of cement paste, mortar, or concrete specimens is usually required as a pre-conditioning step prior to the determination of permeability-related properties according to standard testing methods. The reaction process, and consequently the structure, of an alkali-activated slag or slag/fly ash blend geopolymer binder differs from that of Portland cement, and therefore there is little understanding of the effects of conventional drying methods (as applied to Portland cements) on the structure of the geopolymer binders. Here, oven drying (60 °C), acetone treatment, and desiccator/vacuum drying are applied to sodium silicate-activated slag and slag/fly ash geopolymer pastes after 40 days of curing. Structural characterization via X-ray diffraction, infrared spectroscopy, thermogravimetry, and nitrogen sorption shows that the acetone treatment best preserves the microstructure of the samples, while oven drying modifies the structure of the binding gels, especially in alkali-activated slag paste where it notably changes the pore structure of the binder. This suggests that the pre-conditioned drying of alkali activation-based materials strongly affects their microstructural properties, providing potentially misleading permeability and durability parameters for these materials when pre-conditioned specimens are used during standardized testing.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Standard testing methods for the assessment of the transport properties of cementitious materials require the pre-conditioning (via drying or water removal) of the samples prior to testing to assure that the samples are in a known moisture state. The currently available standard testing methods have been designed for conventional Portland cement-based materials, while no existing standard methods have been specifically developed for the assessment of alkali-activated materials. This has led to the adoption of the Portland cement standards to evaluate activated binders, without questioning the real effects of pre-conditioning or drying on the binder structure, as the microstructural and gel chemical characteristics of activated binders differ from those of Portland cement. Table 1 shows some examples of permeability and durability standard testing methods and the pre-conditioning of the specimens which is required for each test.
Although the standard methods are applied to concrete samples, it is also important to obtain an understanding of the actual behavior of the binder matrix by analyzing paste samples. The analytical techniques used to characterize these microstructural changes are best performed on paste samples to eliminate ambiguity or uncertainty due to aggregates in mortar or concrete specimens.
For conventional Portland cement binders, there is a good understanding of the types of water present in the cement paste, which can be classified according to three types of water environment: pore or capillary water, gel water, and chemically bound water [1, 2]. Capillary water is free to move inside the pores and is not bound by surface forces. Gel water includes adsorbed water and water bound by capillary tension and bonding on the surface of main hydration products such as calcium silicate hydrate (C–S–H) gel. Chemically bound water, also known as non-evaporable water, is held tightly within hydrate phases and can be determined from the decomposition of the phases at temperatures up to 1000 °C. Removal of this water can seriously affect the microstructural characteristics of the material, leading to the physical collapse of hydrated binding phases such as C–S–H.
There have been various studies over the past decade aimed at determining the optimal methods of moisture removal from hydrated Portland cement samples, either intended for pre-conditioning prior to determination of transport properties (in which case partial water removal is generally desirable) or to halt the hydration process so partially reacted samples can be studied by an ex-situ analytical method. Collier et al. [3] report the advantages and disadvantages of drying techniques including oven drying, vacuum drying, solvent replacement, and freeze drying. Their results show that these drying methods do not induce significant structural differences in hardened Portland cement-based pastes and highlight that acetone quenching is the least damaging method. Zhang and Scherer [4] discovered that drying by isopropanol exchange exhibits relatively little effect on the microstructure of cement paste. Other studies of the effects of drying methods on Portland cement pastes have shown that oven drying at 100 °C induces changes in pore size distribution and surface areas as measured by mercury intrusion porosimetry [5, 6] and nitrogen sorption [7]. This was associated with the dehydration and consequent decomposition of the C–S–H-type gel. Solvent replacement techniques using liquids such as methanol and acetone are considered to be gentle with regard to the binder structure, while freeze drying may not completely dry the specimens [2]. During oven drying, the surface tension of the receding water menisci produces stresses that can cause microstructural changes. Drying by replacement of water using solvents such as methanol, ethanol, acetone, or isopropanol is believed to reduce this stress due to the lower surface tension forces as these solvents are removed [8].
Alkali-activated binders, also known as “geopolymers,” are a class of binders that can be based on industrial by-products such as calcined clays, metallurgical slags, and fly ash and have been increasingly investigated in the past few decades [9–11]. The activation of calcium-rich slags with an alkaline activator promotes rapid dissolution of the precursor and a release of species such as Ca and Al to form a C–(A)–S–H (CaO–Al2O3–SiO2–H2O)-type gel [12–14]. The distinctive microstructure of alkali-activated slag cement can give highly desirable structural properties such as strength and durability [15–18]. On the other hand, in alkali-activated aluminosilicate (geopolymer) binders derived from low-calcium materials such as fly ash and metakaolin, a three-dimensional “N–A–S–H”-type gel (Na2O–Al2O3–SiO2–H2O) has been identified as the main reaction product [19]. The chemical and structural characteristics of the N–A–S–H geopolymer gel provide good resistance to aggressive environments, including high temperatures [20, 21]. The high degree of network cross-linking of the 4-coordinated Si and Al sites in this type of gel is in contrast to the two-dimensional silica chains formed in the C–S–H (CaO–SiO2–H2O)-type gels characteristic of Portland cement hydration products and alkali-activated slag binders. Both, however, can promote similar physical and binding characteristics in the hardened binder product [22].
There is little existing understanding of the water environments in alkali-activated materials. Recent studies assessing the role of water in alkali-activated metakaolin binders [23–27] have identified that in these materials, water plays an important role in the kinetics of dissolution of ionic species and polymerization processes and in hardened products, it can exist as absorbed water inside pores or as condensed hydroxyl groups on the surface of the gel. It is believed that the water does not participate notably in the formation of the main product phases (i.e., does not form a strongly hydrated binder phase similar to Portland cement-based materials) and therefore can easily be removed from the material.
There is also little information in the open literature regarding the role of water in alkali-activated slag and slag/fly ash blends. As in Portland cement binders, it is likely that the application of different drying methods to alkali-activated materials will promote changes in the pore structure and modify the binding phases and consequently will give different results regarding the permeability of alkali-activated materials, which is critical in determination of the potential long-term durability of concretes made from these binders, especially when exposed to aggressive environments.
So, based on this need to understand the effects of water removal using different drying methods, this study focuses on the assessment of the structural changes induced in alkali-activated slag and slag/fly ash blends as a consequence of accelerated drying. Characterization of the dried pastes is conducted through X-ray diffraction, Fourier transform infrared spectroscopy, thermogravimetry, and nitrogen sorption. Undried samples are used as a reference for the examination of the structural changes.
Experimental program
Ground granulated blast furnace slag and fly ash, supplied by Zeobond Pty Ltd (Melbourne, Australia), were used as the main binder precursors. Table 2 shows the chemical composition of these raw materials. A detailed characterization of these precursors is reported elsewhere [28], where minor traces of gypsum were identified in the anhydrous slag, and quartz, mullite, hematite, and maghemite were the main crystalline phases in the unreacted fly ash. Two types of binders were produced in this study: an alkali-activated slag and alkali-activated slag/fly ash pastes with a slag/fly ash 1:1 mass ratio.
The alkali activator used was a solid anhydrous sodium metasilicate with 49.1 wt% SiO2 and 50.9 wt% Na2O, also supplied by Zeobond Pty Ltd. The activator, dosed at 8 % by a mass of binder solids, was dissolved in water and allowed to cool to room temperature prior to preparation of the specimens. A water/binder mass ratio of 0.40 was used to prepare the paste specimens, which were cured in sealed polypropylene tubes at 30 °C for 40 days. After curing, the samples were removed from the tubes, then crushed and dried using the following methods:
-
Drying in a laboratory oven at 60 °C for 24 h.
-
Submerging the crushed samples in acetone for 15 min, then filtering using a vacuum pump.
-
Placing the samples inside a desiccator after crushing, and applying vacuum to a gauge pressure of −85 kPa for 24 h.
After drying, the samples were sealed into centrifuge tubes, which were stored in a desiccator over silica gel.
Before and after drying, paste samples were analyzed by the following:
-
X-ray diffraction (XRD) was conducted using a Bruker D8 Advance instrument with Ni-filtered Cu Kα radiation, a step size of 0.020°, and a count time of 2 s per step.
-
Fourier transform infrared (FTIR) spectroscopy was conducted using a Bruker Tensor 27 spectrometer and scanning 32 times from 4000 to 400 cm−1 at 4 cm−1 resolution, using the KBr pellet method.
-
Thermogravimetric analysis (TGA) was conducted using a PerkinElmer Diamond instrument. Samples were crushed by hand using a mortar and pestle, transferred immediately to an alumina crucible, held under isothermal conditions for 60 min at 40 °C to equilibrate, and then heated 40–1000 °C at 10 °C/min in a nitrogen environment. A sample mass of 35 ± 5 mg was used in the analysis of each specimen. The contribution of carbonation to mass loss was calculated as the mass loss between 500 and 800 °C.
-
Nitrogen sorption analysis was conducted using an ASAP 2010 apparatus (Micromeritics). Porosities and pore size distributions were calculated using the Barrett-Joyner-Hallenda (BJH) method [29] from the adsorption branches, and surface area was calculated by the Brunauer-Emmett-Teller (BET) method [30]. Results were analyzed using the Micromeritics TriStar 3000 analysis software.
It is important to mention that during nitrogen sorption testing, it is essential to rinse and dry the specimens before testing to remove any impurities. This means that the reference samples (undried) assessed in this study were subjected to a “drying process” during the degassing procedure, where vacuum pressure below 100 mTorr was applied at 60 °C for 15 h. Usually, degassing is applied at a higher temperature (e.g., 100–120 °C); however, considering that the main goal of the present study was to understand the changes caused by drying, it was crucial to choose a temperature that minimized the changes in the paste structure, so reliable results could be obtained.
Results and discussion
X-ray diffraction
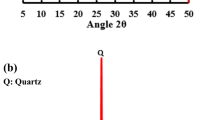
The diffractogram of the undried alkali-activated slag is shown in Fig. 1, along with the specimens dried using the different methods studied. In addition to the disordered binder gel, the main crystalline products identified are a poorly crystalline C–S–H (a structure resembling riversideite, Ca5Si6O16(OH)2, Powder Diffraction File (PDF) # 00-029-0329, but almost certainly with some degree of Al substitution), along with hydrotalcite (Mg6Al2(CO3)(OH)16·4(H2O), PDF# 00-041-1428), consistent with previous studies of activated slag [12, 31–34]. Minor traces of the calcium carbonate polymorphs calcite (CaCO3, PDF# 01-071-3699) and aragonite (CaCO3, PDF# 01-076-0606) are also identified in these samples at similar intensities, regardless of the drying method, and are attributed to atmospheric carbonation of the specimens either before testing or during XRD analysis.

Cu Kα X-ray diffractograms of alkali-activated slag as a function of the drying methods applied. The inset shows difference patterns in the region around the main C–S–H peak, obtained by subtracting the diffractograms as marked and highlighting the changes in this region as a result of drying
Upon drying of the samples in acetone, vacuum, or a 60 °C oven, a slight reduction of the intensity of the main peak attributed to C–S–H (29.5° 2θ) is identified (Fig. 1) when compared with the diffractogram of the undried specimen. The reduction of the C–S–H peak intensity seems to be more notable in vacuum-dried and oven-dried samples (as shown in the inset on Fig. 1), indicating that those drying techniques might be inducing more severe changes in the structure of the hydrated products. However, considering the amorphous nature of the majority of the reaction products formed in activated slag systems, hydrated phases and semi-crystalline features are difficult to distinguish using this technique.
Figure 2 shows the diffractograms of activated slag/fly ash geopolymer samples. In this blended geopolymer system, the main identifiable reaction product is also a poorly crystalline C–S–H-type gel. Minor traces of calcite, along with unreacted fly ash crystalline phases (the quartz, mullite, and hematite previously identified in the precursor), are also observed. Upon drying, slight broadening of the C–S–H peak at 29.5° 2θ is identified via the difference patterns in the inset, along with a minor reduction in its intensity in the vacuum-dried and oven-dried samples, when compared with specimens treated with acetone and the reference (undried) sample. This suggests some structural changes in the C–S–H-type gel, moving toward a less ordered structure than in the untreated specimen. However, the degree of structural change is shown in the inset of Fig. 2 to be much less than in the alkali-activated slag samples.

Cu Kα X-ray diffractograms of alkali-activated slag/fly ash geopolymers as a function of the drying methods applied. The inset shows difference patterns in the region around the main C–S–H peak, obtained by subtracting the diffractograms as marked and highlighting the changes in this region as a result of drying
Fourier transform infrared spectroscopy
A comparison of the FTIR spectra of the dried and undried alkali-activated slag samples is shown in Fig. 3. All spectra show the O–H stretching vibration modes observed between 3200 and 3600 cm−1. The broad peak in this range includes a band at 3450 cm−1 attributed to the asymmetric stretching of H–OH and at 3260 cm−1 attributed to the symmetric stretching of H–OH [35]. The bending mode of H–O–H is also observed at 1640 cm−1. These bands are observed before and after drying of the samples, suggesting that the drying methods do not completely remove the chemically bound water in the hydrated products. However, oven drying at 60 °C and vacuum drying do promote significant changes in the structure of the activated slag. In the vacuum-dried sample, a slight reduction in intensity and a narrowing of the H-OH band centered at 3450 cm−1 are identifiable, compared with the undried and acetone-treated specimens. This indicates that this drying method removes more water from the hydrated products than the acetone method.

Infrared spectra of alkali-activated slag subjected to different drying methods
The band observed at 960 cm−1 in the undried specimen is assigned to the stretching vibration mode of the T–O–T bonds (T: tetrahedral Si or Al) in the C–S–H [36], the intensity of which is significantly reduced in the oven-dried and vacuum-dried specimens. In the region between 900 and 1000 cm−1, three distinctive peaks at 966, 1032, and 1110 cm−1 are identified in the oven-dried samples, and very similar peaks (968, 1032 and 1110 cm−1) are observed in the vacuum-dried samples. The new bands at 1032 and 1110 cm−1 are potentially associated with increased cross-linking of the C–S–H gel during water removal, promoting the formation of a more effectively Si-rich aluminosilicate binding phase when analyzing the bonding environments (as is the case in FTIR). The shoulder at 877 cm−1 is assigned to the asymmetric stretch of AlO4 groups in the Al–O–Si bonds in the binding gel [14, 37], and the fact that it disappears in the vacuum-dried and oven-dried samples indicates disruption in the environment of Al–O–Si bonds in the binding phase.
The spectrum of the acetone-dried sample shows a similar profile to that of the undried sample, although with the removal of more water. The T–O–T band shifts slightly, from 960 to 964 cm−1, after the acetone treatment, which can be associated with the reorganization of the binding gel to compensate for the water removal.
The band at 1420 cm−1 is assigned to the asymmetric stretch modes of the O–C–O bonds in calcite [38], corresponding to the calcite identified in this specimen through X-ray diffraction in Fig. 1. The shoulder near 1060 cm−1 corresponds to the stretching vibration modes of the Al–O and Mg–O bonds in hydrotalcite, and the band at 617 cm−1 is due to the bending of the Al–O and Mg–O bonds of this compound [39].
In the activated slag/fly ash geopolymer (Fig. 4), the bands between 3200 and 3600 cm−1 and 1640 cm−1 assigned to the asymmetric stretching and bending of the H–OH bonds, as previously identified in the activated slag spectra, are also observed. A minor reduction in the intensity of the asymmetric stretching band of H–OH bonds at 3440 cm−1 is observed when comparing the spectra of the acetone-dried and the vacuum-dried samples. The intensities of these bands are again significantly reduced after acetone or oven drying, suggesting the removal of the free water from these samples. Calcite is also observable at 1420–1440 cm−1 in all spectra, consistent with the X-ray diffraction analysis (Fig. 2).

Infrared spectra of alkali-activated slag/fly ash geopolymers subjected to different drying methods
The T–O–T band of the binding phase formed in the undried activated slag/fly ash paste is observed at 1006 cm−1. This band is at a higher wavenumber compared with the activated slag pastes, which is associated with the lower content of calcium in the C–S–H-type product formed in this system and also the formation of a more cross-linked alkali aluminosilicate binding phase as a consequence of the simultaneous activation of fly ash and slag [40, 41]. A broad shoulder near 1100–1166 cm−1 is also observed, assigned to the asymmetric stretching vibrations of Si–O–Si in highly polymerized silicates, corresponding to the unreacted quartz from the fly ash and also overlapping in part with the sodium aluminosilicate (N–A–S–H) gel [42]. The band at 457 cm−1 is attributed to the symmetric stretching of Si–O–Si and O–Si–O bonds in the C–S–H-type gel [36].
After drying, there is a significant shift toward a higher wavenumber of the peak assigned to the C–S–H-type gel. This again suggests reorganization of the gel toward a more cross-linked structure during acetone, vacuum, and oven drying. The shoulders associated with the N–A–S–H gel do not show notable changes upon drying by the methods applied here, suggesting better nanostructural stability upon drying for this type of geopolymer gel. The geopolymer binder system has been reported to withstand high temperature exposure due to the minimal changes in its structure upon release of physically bound water [43]. Although the purpose of that particular study was not specifically to analyze drying of the samples, it did show that a fly ash geopolymer can maintain structural integrity at elevated temperature, suggesting a coherent N–A–S–H structure during drying at the temperature used here. Therefore, it is possible that the C–S–H-type gel is the component of the material that is mainly affected during the drying of the samples.
Thermogravimetric analysis
Tables 3 and 4 show the calculated mass loss for dried and undried activated slag and slag/fly ash samples, determined in different temperature ranges during the thermogravimetry analysis. The tables include the mass loss due to evaporation of free water during 60 min of isothermal hold at 40 °C prior to the start of heating, mass loss during heating from 40 to 1000 °C, and the contribution to mass loss within the temperature range 500–800 °C which is assigned to superficial carbonation of the specimens. The total mass loss of the undried alkali-activated slag pastes (Table 3) is higher than that of slag/fly ash geopolymer pastes (Table 4). However, mass loss during the isothermal hold at 40 °C is higher in the slag/fly ash sample (Table 4), indicating more free water content and less chemically bound water compared to the activated slag pastes. Some of the additional weight loss reported for activated slag samples is attributed to the higher content of reaction products having been formed in these materials at the time of curing at which these samples were assessed, as the slag is more reactive than the fly ash precursor. Chemically bound water is present in the main reaction products C–S–H [44, 45] and hydrotalcite [34], the concentration of which is higher in activated slag pastes compared with activated slag/fly ash geopolymers as identified in XRD. This is indicated in the mass loss between 100 and 300 °C temperature regions, as will be discussed in detail below, where activated slag pastes had a higher mass loss compared to slag/fly ash geopolymer pastes in this temperature range.
Activated slag pastes dried with acetone show (Table 3) a total mass loss which is 8 % lower than undried pastes, the main mass loss of which is taking place during the isothermal period of the thermogravimetry testing. The total mass loss of vacuum-dried and oven-dried activated slag specimens is around half of the mass loss of the undried paste, suggesting that these drying methods are removing not just the free water, but are also promoting the desiccation of the hydrated products and therefore their potential structural collapse. Vacuum drying also has a strong effect on the unbound water as identified by the mass loss during the isothermal period (Table 3). Comparing the mass loss between vacuum and oven drying, it is also shown here that vacuum drying can remove a considerable amount of free water and be damaging to activated slag pastes. These results are consistent with the observations of Gallé [6], who identified that vacuum and oven drying induce stress and a consequent microcracking of the paste. It has been reported [15, 46, 47] that alkali-activated slag binders are subject to self-drying and microcracking if exposed to severe or rapid changes in humidity and temperature, which is in good agreement with this suggestion. There are no significant differences in the mass loss during the heating period among all drying methods tested, and carbonation is not strongly contributing to the mass loss of these specimens.
In alkali-activated slag/fly ash pastes, vacuum and oven drying also promote mass losses of around half of the value reported for this paste before drying (Table 4). This is again consistent with the removal of the water absorbed to the gel pores during drying, along with part of the chemically bound water, which might have a negative effect on the stability of the pore structure of these binders. Even though it is expected that the reaction products forming in activated slag/fly ash binders have water less tightly bonded (i.e., more physically absorbed and less chemically incorporated) to the reaction products, consistent with a more cross-linked structure, it has been identified that in alkali-activated systems based on 1:1 blends of slag and fly ash [41] or slag and metakaolin [48], the structure is mainly dominated by a C–S–H-type binder. On the other hand, reduction of just 2 % in the total mass loss is identified in acetone-dried activated slag/fly ash pastes when compared with undried specimens, suggesting that this drying method is not inducing significant structural changes in the binding phases in this system.
Differential thermograms of alkali-activated slag paste (Fig. 5) show the changes in the intensity of the low-temperature peak attributed to the loosely bound water before and after drying. The first mass loss peak is assigned to the release of evaporable water in the reaction products, in particular C–S–H. In conventional Portland cement systems, evaporable water is completely removed by 120 °C [49]; however, this may differ in activated slag binders due to the differences in chemistry and the higher degree of cross-linking of the binding phase. In the undried alkali-activated slag paste, the first peak, consistent with the release of water from the C–S–H gel, is observed at 108 °C. The intensity of this peak markedly decreases when the samples are dried, consistent with the reduced total mass loss identified in these samples (Table 3).

Differential thermograms (mass loss downward) of alkali-activated slag with each drying method applied. The isothermal hold period is not shown
Acetone drying of the activated slag paste induces a shift of the first peak toward higher temperatures (116 °C), consistent with the removal of free water by the acetone treatment and the dehydration of the more tightly chemically bonded water present in the C–S–H phase. In vacuum-dried and oven-dried specimens, the first dehydration peak is shifted to 135 °C, indicating that during the drying of the pastes using these two methods, both free water and chemically bonded water are removed from the C–S–H-type gel, which induces the structural changes in the binding phases as observed through FTIR (Fig. 4).
The mass loss between 210 and 300 °C identified in all samples assessed corresponds to the dehydration of hydrotalcite [34]. Decomposition of calcium carbonate is observed between 500 and 800 °C, consistent with the identification by X-ray diffraction (Fig. 1) of calcite and aragonite.
The undried slag/fly ash geopolymer (Fig. 6) also exhibits a peak at 105 °C, attributed to the dehydration of the C–A–S–H-type gel and to the freely evaporable water present in large pores in the geopolymer gel formed by the activation of the fly ash. Mass loss between 250 and 270 °C is again assigned to the dehydration of hydrotalcite, as identified in this binder through X-ray diffraction (Fig. 2).

Differential thermograms (mass loss downward) of alkali-activated slag/fly ash geopolymer with each drying method applied. The isothermal hold period is not shown
The drying methods applied to this paste also induce a reduction in intensity and shift toward higher temperatures of the first peak assigned to the dehydration of the reaction products, along with the broadening of this region. This indicates that the drying of these specimens is also inducing structural changes in the binding phases. Oven drying and vacuum drying lead to the shifting of the first peak to 134 and 147 °C, respectively, suggesting that these drying methods might be removing some of the water absorbed in the N–A–S–H-type gel (“zeolitic water”) and are potentially destroying the structure of the C–S–H-type gel in this system, as the dehydration of this phase is expected at temperatures below 120 °C. The shift of the first peak toward higher temperatures is consistent with the formation of a structure containing zeolitic-like water environments, which is in agreement with the identification through FTIR of a binding gel with an increased degree of cross-linking in the dried specimens. Calcium carbonate decomposition can be observed between 500 and 640 °C. These results indicate that the inclusion of fly ash in activated slag binders induces the formation of reaction products with water more tightly bonded and with a different pore structure than in the activated slag binders.
Nitrogen sorption
The N2 sorption–desorption isotherms for all of the pastes tested, before and after drying, show a type V classification with a hysteresis loop, characteristic of solids having micropores (up to 2 nm) or mesopores (2–50 nm), with an inflection point at a fairly high relative pressure [2]. Table 5 compares the BETN2 surface areas of both alkali-activated slag and alkali-activated slag/fly ash geopolymer samples as calculated from these isotherms, where it can be seen that the undried activated slag/fly ash geopolymer has a higher surface area than alkali-activated slag, consistent with the more porous nature of the siliceous materials comprising the binder in the blended slag/fly ash sample [41]. In hydrated Portland cements, the BETN2 surface area varies from 10 to 200 m2/g, depending on factors such as water/cement ratio, drying techniques, and humidity [50–52], and so the values obtained in this study are within the range expected for conventional cement binders.
Specimens dried in an oven at 60 °C report the lowest surface areas among those analyzed, for both activated slag and slag/fly ash samples. This is consistent with other studies reporting [7, 53] that oven drying decreases the total pore volume of cement paste, as the collapse of pores is promoted by the stresses induced through the desiccation of the reaction products, and therefore the BETN2 surface area is reduced. A small change in the surface area of the pastes is identified in vacuum-dried activated slag specimens, which differs from the structural changes identified in these specimens through other analytical techniques. It is likely that the degassing process (which involves holding the specimens at 60 °C under vacuum for 15 h) required for the nitrogen sorption testing is affecting the “un-dried” samples, so that similar structural changes to those induced during vacuum drying are taking place in the control specimens in this test. However, the acetone-dried slag paste does show a reduction in the BETN2 values compared with the undried specimen. According to the FTIR and XRD results discussed above, acetone drying does not induce structural changes in this binder; however, thermogravimetry results (Table 3) show that this method removes large amounts of free water from the sample, which is likely to open pores, making the paste susceptible to pore collapse via further desiccation during the degassing process of the BETN2 test procedure.
For the blended slag/fly ash geopolymer sample, comparable BETN2 values are obtained using each of the different drying methods assessed, except for oven drying which seems to induce a substantial reduction of the specific surface area, as was also identified in alkali-activated slag samples. In this case, acetone-dried and vacuum-dried pastes exhibit very similar measured surface areas to the undried paste, indicating that these drying methods preserve the pore structure of the material, while the oven-dried paste shows a significant reduction in surface area, associated with the collapse of the binding phase structure as discussed above. Thermogravimetry data for the slag/fly ash binders (Fig. 6) revealed structural damage to the C–A–S–H-type gel after oven drying, along with the dehydration of the geopolymer gel, which is consistent with the remarkable difference in the surface area of the oven-dried specimen when compared with undried paste. The smallest pores will undergo more deformation than the larger pores because the smallest pores are most likely to collapse during drying due to the increase in capillary pressure with decreasing pore size [7], which leads to a loss of the surface area which would usually have been contributed by these pores.
For Portland cement binders, it has been reported [7] that during acetone drying, the water in the pores is replaced by acetone with a lower surface tension, reducing capillary pressure during drying and preventing pore collapse. From the data obtained here, it appears that this is the case for the blended slag/fly ash geopolymer, but not for the alkali-activated slag samples. This may be related to differences in bound water content and/or pore radius between the two types of alkali-activated binder.
Further analysis of the nitrogen sorption isotherms was conducted via pore size distribution calculations in accordance with the Barrett-Joyner-Halenda (BJH) method [29]. Figures 7 and 8 show the differential pore size distributions for alkali-activated slag and activated slag/fly ash pastes, respectively, calculated from the adsorption branch of the isotherm. The adsorption branch is chosen to plot the pore size distribution to overcome uncertainty due to the portion of the fluid that is unable to evaporate during desorption due to the presence of narrow ink bottle-shaped pores in these materials [2, 54, 55].

BJH pore size distributions calculated for alkali-activated slag before and after drying

BJH pore size distributions calculated for alkali-activated slag/fly ash geopolymers before and after drying
Alkali-activated slag pastes (Fig. 7) show pore sizes spanning the full range measurable by this technique (1.8–200 nm), corresponding to the range from mesopores to macropores, and it is evident that the drying methods are inducing changes in the pore size distribution. A reduced pore volume is observed in every dried paste when compared with the undried control. Oven drying promotes the reduction of the mesopore volume, which is consistent with the high quantities of water removed in this drying method as observed through thermogravimetry (Fig. 5). Desiccation of the samples leads to the widening of the gel pores, accompanied by their partial collapse and a consequent microcracking of the specimens [56]. There seems to be a high susceptibility to drying-induced structural changes in the C–S–H gel phase, and specifically in the gel pore network, formed in alkali-activated slag binders.
Vacuum drying seems to have a different effect on the pore size distribution when compared with oven drying. In this case, the concentration of smaller pores seems to be comparable to what is observed in undried samples, but there is a remarkable reduction in the volume of large pores in the sample, suggesting that this drying method is somehow inducing the refinement of the pore structure, most likely through the contraction of the binding gel. Conversely, acetone-dried alkali-activated slag paste seems to present a reduced concentration of both small and large pores compared with the undried sample, suggesting that the water replacement by acetone has partially dehydrated the C–S–H gels [57]. These results suggest that vacuum and acetone drying remove the water present in the large pores, leading to collapse of these pores and reduction in their volume in alkali-activated slag binders.
Based on the BJH pore size distribution of the alkali-activated slag/fly ash geopolymers (Fig. 8), two porosity regions are apparent, micropores (1.7–8 nm) and mesopores (10–100 nm) with a bimodal distribution. Drying at 60 °C leads to a dramatic change in the pore size distribution of alkali-activated slag/fly ash binders, compared with undried paste, as the micropores are no longer observed in the sample and there is a remarkable reduction in the total pore volume. This indicates that oven drying of these pastes has the most detrimental effect in terms of modifying the pore structure of the paste among the drying methods assessed.
Vacuum and acetone drying seem to each induce a slight increment in the concentration of pore volume in the micropore region in the geopolymer samples, with a consequent reduction in the pore volume in the mesopore region; however, the pore size distributions of samples dried using these two methods are similar to those obtained in undried samples, consistent with the limited extent of structural changes identified in these samples through FTIR (Fig. 4) and DTG (Fig. 6). The binding phase formed in the slag/fly ash geopolymer seems to be less prone to structural changes than the binding gel formed when slag is solely used as precursor, as it can be expected that the water is chemically bonded in a different environment in each case. In particular, the more cross-linked gel in the blended slag/fly ash geopolymer is more resistant to structural collapse when dried at ambient temperature because its water is predominantly physically, rather than chemically, bound.
Selection of drying methods
Based on the outcomes of this work, we propose that when it is necessary to dry specimens of alkali-activated pastes, the following should be taken into consideration:
-
When the material contains a significant content of chemically bound water (i.e., alkali-activated slag), drying should be achieved by the acetone method.
-
When the material is low in chemically bound water (i.e., binders based on fly ash or metakaolin), drying should be achieved by either vacuum or acetone methods.
These recommendations are based on the concept that it is desirable, as far as possible, to avoid alteration of the binder gel and/or pore structures as a result of the drying process. This also leads to the suggestion that many commonly used testing protocols involve the application of sample pre-conditioning conditions which are inappropriate for alkali-activated binder materials and may be causing significant damage to the samples prior to analysis.
It is also notable that there is a significant difference between the conditions imposed by the standard drying methods and actual field conditions because drying in the laboratory is much more rapid and so induces a higher degree of mechanical stress within the gels. The drying methods adopted in these standard tests are based on the understanding that water removal to a specific moisture state is needed when assessing the durability of different materials, whereas in actual service, drying at such high temperatures or low water vapor partial pressures is not experienced. Therefore, the drying methods used are much harsher than service conditions, meaning that the results obtained here should not be extrapolated to predict performance of AAMs under natural wet–dry cycling exposure, where the changes in moisture state are much slower.
Conclusions
An assessment of the effects of different drying methods on the structure of alkali-activated slag and blended slag/fly ash geopolymer pastes shows that the tendency of each method to induce structural changes in the paste is strongly dependent on the binder gel structure. Alkali-activated slag pastes show structural changes in the binding gel under vacuum drying or oven drying at 60 °C, associated with desiccation and chemical changes in the C–S–H-type gel. However, different results are obtained in activated slag/fly ash geopolymer paste, which does not exhibit notable chemical structural changes after drying by the different methods tested, although oven drying does still lead to some degree of pore collapse. This difference between sample types is related to the mode of water binding in the gel structures, where the aggressive removal of water of hydration from C–S–H-type gels is more damaging than the removal of physically bound (similar to zeolitic) water from alkali aluminosilicate gels. Solvent replacement using acetone appears to be the gentlest drying method to remove free water from the binders, while preserving the structural features of the binding gels and the pore network. These results highlight the need to reassess the drying procedures adopted in many standard permeability and durability testing methods when applied to alkali-activated materials, as sample pre-conditioning is likely to be inducing structural changes in the material that can potentially give misleading conclusions regarding the long-term in-service performance of these binders.
References
Mehta PK, Monteiro PJM (2006) Concrete: microstructure properties and materials, 3rd edn. McGraw-Hill, New York
Aligizaki KK (2006) Pore structure of cement-based materials; testing, interpretation and requirements. Taylor & Francis, New York
Collier NC, Sharp JH, Milestone NB, Hill J, Godfrey IH (2008) Cem Concr Res 38:737
Zhang J, Scherer GW (2011) Cem Concr Res 41:1024
Moukwa M, Aitcin PC (1988) Cem Concr Res 18:745
Gallé C (2001) Cem Concr Res 31:1467
Juenger MC, Jennings HM (2001) Cem Concr Res 31:883
Day RL (1981) Cem Concr Res 11:341
Buchwald A, Schulz M (2005) Cem Concr Res 35:968
Provis JL, van Deventer JSJ (2009) Geopolymers: structure, processing, properties and industrial applications. Woodhead Publishing Limited, UK
van Deventer JSJ, Provis JL, Duxson P, Brice DG (2010) Waste Biomass Valoris 1:145
Brough AR, Atkinson A (2002) Cem Concr Res 32:865
Fernández-Jiménez A, Puertas F (2003) J Am Ceram Soc 86:1389
Bernal SA, Provis JL, Mejía de Gutierrez R, Rose V (2011) Cem Concr Compos 33:46
Bernal SA, Mejía de Gutierrez R, Pedraza AL, Provis JL, Rodríguez ED, Delvasto S (2011) Cem Concr Res 41:1
Roy DM, Jiang W, Silsbee MR (2000) Cem Concr Res 30:1879
Bakharev T, Sanjayan JG, Cheng YB (2002) Cem Concr Res 32:211
Bernal SA, Mejía de Gutiérrez R, Provis JL (2012) Constr Build Mater 33:99
Fernández-Jiménez A, Palomo A, Criado M (2005) Cem Concr Res 35:1204
Duxson P, Lukey GC, van Deventer JSJ (2006) J Non-Cryst Solids 352:5541
Provis JL, Yong CZ, Duxson P, van Deventer JSJ (2009) Colloids Surf A 336:57
Fernández-Jiménez A, Palomo A (2005) Cem Concr Res 35:1984
Provis JL, van Deventer JSJ (2007) Chem Eng Sci 62:2318
Duxson P, Lukey GC, van Deventer JSJ (2007) J Non-Cryst Solids 353:2186
Steins P, Poulesquen A, Diat O, Frizon F (2012) Langmuir 28:8502
Zuhua Z, Xiao Y, Huajun Z, Yue C (2009) Appl Clay Sci 43:218
Lizcano M, Gonzalez A, Basu S, Lozano K, Radovic M (2012) J Am Ceram Soc 95:2169
Ismail I, Bernal SA, Provis JL, Hamdan S, van Deventer JSJ (2013) Mater Struct. doi:10.1617/s11527-012-9906-2
Barrett EP, Joyner LG, Halenda PP (1951) J Am Chem Soc 73:373
Brunauer S, Emmett PH, Teller E (1938) J Am Chem Soc 60:309
Bakharev T, Sanjayan JG, Cheng Y-B (1999) Cem Concr Res 29:113
Wang SD, Scrivener KL (1995) Cem Concr Res 25:561
Oh JE, Monteiro PJM, Jun SS, Choi S, Clark SM (2011) Cem Concr Res 40:189
Ben Haha M, Lothenbach B, Le Saout G, Winnefeld F (2011) Cem Concr Res 41:955
Farmer VC (1974) The infrared spectra of minerals. Mineralogical Society, London
García-Lodeiro I, Fernández-Jiménez A, Blanco MT, Palomo A (2008) J Sol–Gel Sci Technol 45:63
Puertas F, Fernández-Jiménez A (2003) Cem Concr Compos 25:287
Gadsden JA (1975) Infrared spectra of minerals and related inorganic compounds. Butterworths, London
López T, Bosch P, Asomoza M, Gómez R, Ramos E (1997) Mater Lett 31:311
Puertas F, Martínez-Ramírez S, Alonso S, Vázquez E (2000) Cem Concr Res 30:1625
Provis JL, Myers RJ, White CE, Rose V, van Deventer JSJ (2012) Cem Concr Res 42:855
Criado M, Fernández-Jiménez A, Palomo A (2007) Microporous Mesoporous Mater 106:180
Kong DLY, Sanjayan JG, Sagoe-Crentsil K (2007) Cem Concr Res 37:1583
El-Jazairi B, Illston JM (1977) Cem Concr Res 7:247
Girão AV, Richardson IG, Porteneuve CB, Brydson RMD (2007) Cem Concr Res 37:1571
Collins F, Sanjayan JG (2001) Cem Concr Compos 23:345
Collins F, Sanjayan JG (2000) Cem Concr Res 30:791
Bernal SA, Rodríguez ED, Mejía de Gutierrez R, Gordillo M, Provis JL (2011) J Mater Sci 46:5477. doi:10.1007/s10853-011-5490-z
Alarcon-Ruiz L, Platret G, Massieu E, Ehrlacher A (2005) Cem Concr Res 35:609
Thomas JJ, Hsieh J, Jennings HM (1996) Adv Cem Based Mater 3:76
Litvan GG (1976) Cem Concr Res 6:139
Litvan GG, Myers RE (1983) Cem Concr Res 13:49
Sarawade PB, Kim J-K, Hilonga A, Quang DV, Kim HT (2011) Appl Surf Sci 258:955
Lowell S, Shields JE, Thomas MA, Matthias T (2004) Characterization of porous solids and powders : surface area, pore size and density. particle technology series. Kluwer Academic Publishers, Dordrecht
Lloyd RR, Provis JL, Smeaton KJ, van Deventer JSJ (2009) Microporous Mesoporous Mater 126:32
Korpa A, Trettin R (2006) Cem Concr Res 36:634
Mitchell LD, Margeson JC (2006) J Therm Anal Calorim 86:591
American Society for Testing and Materials ASTM C642 (2006) Standard test method for density, absorption, and voids in hardened concrete
American Society for Testing and Materials ASTM C1585 (2011) Standard test method for measurement of rate of absorption of water by hydraulic cement concretes
Australian Standard AS 1012 (1999) Methods of Testing Concrete Method 21: Determination of water absorption and apparent volume of permeable voids in hardened concrete
Nordtest Method NT Build 492 (1999) Chloride migration coefficient from non-steady state migration experiments
RILEM TC 116-PCD (1999) Mater Struct 32:174
Acknowledgements
The authors acknowledge the Malaysian Ministry of Higher Education for the funding of this project through the Melbourne-Malaysia Split Ph.D. program. This study has also been supported by the Australian Research Council, through a Linkage Project grant cosponsored by Zeobond Research and via the Discovery Grants program, including some support through the Particulate Fluids Processing Centre.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ismail, I., Bernal, S.A., Provis, J.L. et al. Drying-induced changes in the structure of alkali-activated pastes. J Mater Sci 48, 3566–3577 (2013). https://doi.org/10.1007/s10853-013-7152-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7152-9