
Abstract
We report measurements of the X-ray diffraction and X-ray photoelectron spectrum on single crystals of Ag2In2GeS6. We also present first principles calculations of the band structure and density of states using the state-of-the-art full potential augmented plane wave method with different possible approximation for the exchange correlation potential. In this paper, we make a detailed comparison of the density of states deduced from the X-ray photoelectron spectra with our calculations. The theoretical results of the density of states are in reasonable agreement with the X-ray photoelectron spectroscopy (VB-XPS) measurements with respect to peak positions. The calculated density of states shows there is a strong hybridization between the states in the valence and conduction bands states. We have calculated the electron charge density distribution in the (100) and (110) planes. In the plane (100), there exists Ag, In, and S atoms, while the plane (110) Ag, S, and Ge atoms are present. The bonding properties are obtained from the charge density distributions. The calculation show that there is partial ionic and strong covalent bonding between Ag–S, In–S, and Ge–S atoms depending on Pauling electro-negativity difference of S (2.58), Ge (2.01), Ag (1.93), and In (31.78) atoms.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A chalcogenide is a chemical compound consisting of at least one chalcogen element and at least one electropositive element. Although all group 16 elements of the periodic table are defined as chalcogens, the term is more commonly reserved for sulfides, selenides, and tellurides, rather than oxides. The chalcogenide semiconducting compounds have received much attention in recent years due to their possible applications as materials for visible-infrared spectral range [1–7]. They form a large group of semiconducting materials with diverse optical, electrical, structural properties. The band gap is formed from p-chalcogenide delocalized states in the valance band and by relatively more localized s- and d-cationic states in the conduction band [8]. The accentricity of the crystal structure in the chalcogenides is one of the reasons for their possible application as non-linear optical materials [9]. Recently, Chmiel et al. [10] presented complex spectral studies of near-band gap and photoconductive spectra for novel Ag2In2SiS6 and Ag2In2GeS6 single crystals. The spectral dependences of photoconductivity clearly show an existence of spectral maxima within the 450–540 and 780–920 nm. The fundamental absorption edge is analyzed by Urbach rule. The origin of the spectral photoconductivity spectral maxima is discussed. Temperature dependences of the spectra were done. The obtained spectral features allow proposing these crystals as photosensors. An analysis of the absorption and photoconductivity spectra is given within a framework of an oversimplified spectroscopic model of complex chalcogenide crystals. Yet up to now, there is no comprehensive work that concerns the electronic structure of the Ag2In2GeS6 compound. Therefore, it is worthwhile to perform ab initio calculations using a full potential method. In the last decade, ab initio calculations have been successfully used to obtain electronic properties of materials. The structural parameters and dynamical properties of ternary and quarterly crystals determine a wide range of microscopic and macroscopic behavior: photoinduced changes, nonlinear optics, IR-stimulated piezoelectrics, interesting infrared spectra infrared absorption, inelastic neutron scattering, specific heat, etc., [6–14].
In this paper, we present a combined experimental-theoretical study of Ag2In2GeS6.
We report measurements of the X-ray diffraction (XRD), and X-ray photoelectron (XPS) spectrum on single crystals of Ag2In2GeS6. The XRD data gives the positions of the atoms in the unit cell. These are used to perform density functional calculations which optimize the atomic positions so as to give almost zero force on the atoms. This optimized structure is used to calculate the electronic properties such as band structure, density of states, charge density, etc. Calculations are performed using four different exchange correlation potentials to ascertain how these change the electronic properties. Our calculated density of states is compared with the XPS data. We also analyze the charge density so as to determine the nature of the bonding in this compound. The nature of the angular momentum character of the bands suggests the origin of the band gap. We feel that our work can be used for band engineering in future. As there are no calculations for this compound, we thought it worthwhile to do electronic structure calculations.
Structural properties and computational details
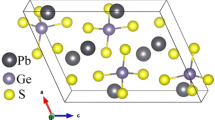
The quaternary sulfides Ag2In2GeS6 crystallize in the monoclinic, Cc space group. The lattice parameters a = 12.2088 Å, b = 12.1978 Å, c = 7.2114 Å, and β is 109.508°. Figure 1 depicts space location of sulfur polyhedral surrounding Ag-(a), In-(b), and Ge-(c) for the optimized structure. In the presented structure, polyhedron surrounding the atoms does not change their shape and the form of a tetrahedron. Following Table 2, one can see that for the polyhedra chemical bond experimental distances are generally enhanced and the final polyhedral have more perfect shape. Using the X-ray diffraction data (XRD), we have optimized the structure by minimization of the forces acting on the atoms. The structure is fully relaxed until the forces on the atoms reach values less than 1 mRy/a.u. The optimized atomic positions along with those obtained from XRD are listed in Table 1. Once the forces are minimized in this construction, one can then find the self-consistent density at these positions by turning off the relaxations and driving the system to self-consistency. From the obtained relaxed geometry, the electronic structure and the chemical bonding can be determined and various spectroscopic features can be simulated and compared with experimental data.

Location of sulfur atomic polyhedral surrounding metallic atoms Ag (a), In (b), and Ge (c) for optimized model structure (atoms belonging to polyhedral remarked)
The all-electron full potential linearized augmented plane wave (FP-LAPW) method was used to solve the Kohn–Sham DFT equations within the framework of the WIEN2K code [15]. This is an implementation of the DFT [16] with different possible approximation for the exchange correlation (XC) potentials. We have employed the local density approximation (LDA) by Ceperley–Alder (CA) [17], and the gradient approximation (GGA) [18], which are based on exchange–correlation energy optimization to calculate the total energy. In addition, Engel–Vosko generalized gradient approximation (EV-GGA) [19], and modified Becke–Johnson potential (mBJ) [20] were also used to avoid the well-known LDA and GGA underestimation of the band gaps. Our calculations demonstrate the effect of the four different kinds of exchange–correlation potentials on the electronic structure and hence the band gap.
The unit cell was divided into two regions, the spherical harmonic expansion was used inside the non-overlapping spheres of muffin-tin radius (R MT) and the plane wave basis set was chosen in the interstitial region (IR) of the unit cell.
The R MT for Ag, In, Ge, and S were chosen in such a way that the spheres did not overlap. In order to get the total energy convergence, the basis functions in the IR were expanded up to R MT × K max = 7.0 and inside the atomic spheres for the wave function. The maximum value of l were taken as l max = 10, while the charge density is Fourier expanded up to G max = 20 (a u)−1. We have used 35 k-points in the irreducible Brillouin zone for structural optimization. For calculating the total and the angular momentum decomposition of the atoms projected electronic density of states, a denser mesh of 290 k-points was used. The convergence of the total energy in the self-consistent calculations is taken with respect to the total charge of the system with a tolerance 0.0001 electron charges.
Results and discussion
The electronic band structure dispersion in k-space along the high symmetry directions in the irreducible Brillouin zone, the total and the angular momentum decomposition of the atoms projected electronic density of states for Ag2In2GeS6 compound are shown in Figs. 2 and 3. Our calculations demonstrate the effect of using four different kinds of exchange–correlation potentials (LDA, GGA, EVGGA, and mBJ) on the electronic structure and hence the band gap (see Fig. 3a). In all cases, the valence band maximum (VBM) and the conduction band minimum (CBM) are located at \( \Upgamma \) resulting in a direct energy gap of 0.67, 0.85. 1.53, and 1.88 eV for LDA, GGA, EVGGA, and mBJ respectively (see Figs. 2, 3a). We should emphasize that mBJ brings the band gap close to our measured one (1.95 eV). It is clear that the difference between the calculated and measured gap is just 0.07 eV, which is an excellent agreement between the experiment and DFT calculation that is attributed to the use of mBJ with full potential. In full potential methods, the potential and charge density are expanded into lattice harmonics inside each atomic sphere and as a Fourier series in the interstitial region. The band gap is formed from p-chalcogenide delocalized states in the valence band and by relatively more localized s and d-cationic states in the conduction band. The increase in the energy gap when move from LDA to mBJ can be attributed due to the fact that the conduction band minimum has strong cation-s states whereas the other states in the conduction band are more heavily mixed with other atomic orbitals such as anion-p states. In the conduction bands shifting, the Ag-s states has small effect and the In-s states has a strong effect in opening the gap, while leaving the valence bands unchanged. Hence, the conduction bands shift toward higher energies with respect to Fermi energy (E F) when move from LDA to GGA, to EVGGA then mBJ. In general, the overall increase in gap is consistent with an overall strengthen of the bonds and, therefore, with a greater bonding antibonding splitting.

The calculated band structure. a LDA, b GGA. c EVGGA. d mBJ

Calculated total and partial densities of states (states/eV unit cell)
The band structure and the density of states can be divided into eight groups/structures. From the angular momentum decomposition of the atoms projected electronic density of states, one can identify the angular momentum character of the various structures. The lowest energy group around −15.0 eV has mainly In-d states. The second group between −14.0 and −13.0 eV has significant contributions from Ge-s, S-s states with small contribution from S-p states. The third group/structure around −12.0 eV is mainly from S-s, In-s/p, Ge-s/p/d, and Ag-p states. The fourth group/structure which is situated between −8.0 and −7.0 eV, is dominated by Ge-s, S-s/p, and In-s states. Admixture of In-s/p and S-s/p states with small contribution of Ge-s/p/d and Ag-s/p states are formed the fifth structure around −5.0 eV. The energy region extended from −4.5 eV up to Fermi energy is forming the sixth structure which is mainly dominated by Ag-d, S-p, Ge-p, In-p, and Ag-s/p states with small contribution of Ge-d states. The electronic structure of the upper valence band is dominated by the Ag-d and S-p interactions. It is clear that all Ag-d character is concentrated in the upper valence band, with only negligible amounts in the conduction band. Most of the Ag-s states are pushed from the valence bands into conduction bands. Finally, the energy region starting from CBM and above results in the seventh and eighth structures which are due to In-s, Ge-s, and S-p states with small contributions from S-s, Ge-p, In-p, and Ag-s/p states.
Deeply looking into the angular momentum decomposition of the atoms projected electronic density of states for Ag2In2GeS6 compound, one can see there is a strong hybridization between Ge-s and S-s at around −13.0 eV, S-p hybridizes with Ge-s and In-s around −12.0 eV. Ge-s and S-p strongly hybridize with S-s around −6.0 eV. Both of In-s and S-p states are hybridized strongly around −4.0 eV. At around −5.0 eV In-s hybridizes with both of S-s/p states, at the same region Ge-p hybridizes with Ag-s/p states. At the CBM Ag-s/p, In-s/p, S-p, and Ge-s states are strongly hybridize with each others. Ge-p state hybridizes with In-p and Ag-s states at 4.0 eV. At 5.0 eV Ge-s and In-s hybridizes with S-s state. The states Ag-s/p hybridize with In-p and Ge-p states around 7.0 eV.
In Fig. 4, our experimental VB-XPS data of Ag2In2GeS6 compound is compared with results of our full potential linear augmented plane wave method using mBJ exchange correlation functional. The calculated TDOS spectrum reproduces the general structure of the measured XPS valence band correctly which is probably attributed to the better representation of the wave functions in the FP-LAPW method. The valence band density of states data was analyzed in the energy range in which the experiment was performed (−8.0–0.0 eV). We note that a reasonable agreement in the matter of the general behavior and position of the peaks. However, the theoretical DOS show more peaks (structures) than the experimental XPS due to the use of small broadening. In the present calculation, the broadening is around 0.003 eV. The most intensive lines in the range −1.5 to −4.5 eV mainly correspond to Ag-d states, while the ones below that value correspond to Ge-s/p, In-s, and S-p states. From comparing our results of the measured VB-XPS with the calculated total densities of states and the angular momentum projected densities of states, we can elucidate the origin of chemical bonding following the same method we used in our previous work [21]. We observe that the VB-XPS, TDOS, and PDOS ranging from −8.0 eV up to Fermi energy (E F) are larger for Ag-d states (5.8 e/eV), Ge-s states (1.1 e/eV), S-p states (0.9 e/eV), and In-s states (0.8 e/eV). One can conclude that some electrons from Ag, In, Ge, and S atoms are transferred into valence bands and contribute in covalence interactions between In–S, Ge–S, and Ag–S atoms. The interaction of charges between In–S, Ge–S, and Ag–S atoms is due to the strong hybridization, and the covalent bond arises due to the degree of hybridization. Hence, there is a strong covalent bonding between these atoms. The angular momentum decomposition of the atoms projected density of states help to analyze the nature of the bonds according to a classical chemical concept. This concept is very useful to classify compounds into different categories with respect to different chemical and physical properties. To support this statement, we have taken a more careful look at the bonding situation since the existence of real hybridization between states of atoms should lead to covalent bond’s origin between these atoms. We have calculated the bond lengths and angles, and good agreement was found between our calculated bond lengths and angles with our measured ones see Tables 2 and 3. Following the Table 2, the maximal difference (δ |exp-optim|) for inter-atomic distances obtained from experiment and optimization cation–anion are observed for atoms Ag(2) and Ge(1) for which in the Table 3 angles between the atoms in the nearest coordination surroundings are presented.

Calculated total density of states using mBJ-FPLAPW method in comparison with the measured total density of states using XPS technique for the valence band
We have calculated the electron charge density distribution in the unit cell in different crystallographic planes. The electronic charge density contour is illustrated in (100) and (110) planes. In the plane (100), there exists only the Ag, In, and S atoms, while the plane (110) the Ag, S, and Ge atoms are present. These planes show that here exists a considerable anisotropy. These two planes are illustrated in Fig. 5a, b. Following these figures, the chemical bonding features can be analyzed. It is clear that there is partial ionic and strong covalent bonding between Ag–S, In–S, and Ge–S atoms depending on Pauling electro-negativity difference of S (2.58), Ge (2.01), Ag (1.93), and In (31.78) atoms. These contour plots show that the majority of Ag, In, and Ge electronic charge is transferred to S atom. One can see that the S atoms are more electronegative than Ag, In, and Ge atoms, and more charge accumulates near S along the bonds and the charge around S atoms are uniformly distributed. This can be seen easily by color charge density scale where blue color (+1.00) corresponds to the maximum charge accumulation site. The distribution of electronic charge around Ag, In, Ge, and S atoms is spherical. While it is non-spherical around Ag–S, Ge–S, and In–S atoms which results in the bonding between these atoms showing prevailingly partial ionic and strong covalent features because the small electro-negativity difference between Ag–S (0.65), Ge–S (0.57), and In–S (0.8).

Electronic space charge density distribution contour calculated with mBJ in the; a (100) plane; b (110) plane
Conclusions
The all-electron full potential linearized augmented plane wave method was used to solve the Kohn–Sham DFT equations within the framework of the WIEN2K code. We have employed different approximation for the exchange correlation potentials namely; LDA, GGA, EVGGA, and mBJ. EVGGA and mBJ were used to avoid the well-known LDA and GGA underestimation of the band gaps. Our calculations demonstrate the effect of the four different kinds of exchange–correlation potentials on the electronic structure and hence the band gap. X-ray diffraction data and X-ray photoelectron spectrum on single crystals of Ag2In2GeS6 were reported. A detailed comparison of DOS-VB deduced from the X-ray photoelectron spectra with our calculated DOS shows reasonable agreement. From the partial density of states a strong/weak hybridization between the states in the valence and conduction bands was observed. We have calculated the bond lengths and angles, good agreement was found between our calculated bond lengths and angles with our measured ones. The maximal difference (δ |exp-optim|) for inter-atomic distances obtained from experiment and optimization cation–anion were observed for atoms Ag(2) and Ge(1) for which in the Table 3 angles between the atoms in the nearest coordination surroundings are presented.
References;
Lekse W, Moreau MA, McNerny KL, Yeon J, Halasyamani PS, Aitken JA (2009) Inorg Chem 48:7516
Lekse JW, Leverett BM, Lake CH, Aitken JA (2008) J Solid State Chem 181
Matsushita H, Katsui A (2005) J Phys Chem Solids 66:1933
Todorov TK, Reuter KB, Mitzi DB (2010) Adv Mater 22:E156
Zhou Z, Wang Y, Xu D, Zhang Y (2010) Sol Energy Mater Sol Cells 94:2042
Davydyuk GE, Myronchuk GL, Kityk IV, Danyl’chuk SP, Bozhko VV, Parasyuk OV (2011) Opt Mater 33:1302
Parasyuk OV, Piskach LV, Romanyuk YE, Olekseyuk ID, Zaremba VI, Pekhnyo VI (2005) J Alloys Compd 397:85
Fedorchuk AO, Gorgut GP, Parasyuk OV, Lakshminarayana G, Kityk IV, Piasecki M (2011) J Phys Chem Solids 72:1354
Sachanyuk VP, Gorgut GP, Atuchin VV, Olekseyuk ID, Parasyuk OV (2008) J Alloys Compd 452:348
Chmiel M, Piasecki M, Myronchuk G, Lakshminarayana G, Reshak Ali H, Parasyuk OG, Kogut Yu, Kityk IV (2012) Spectrochim Acta A 91:48
Dovgii Ya O, Kityk IV (1991) Phys Stat Sol B 166:395
Dovgii YO, Kityk IV (1991) Soviet Phys Semicond-USSR 25(10):1108
Dovgii YO, Kityk IV (1991) Kristallograiya 36(3):772
Dovgii YO, Kityk IV, Mankovskaya IG (1990) Fizika Tverdogo Tela 32(10):3170
Blaha P, Schwarz K, Madsen GKH, Kvasnicka D, Luitz J (2001) WIEN2K, an augmented plane wave + local orbitals program for calculating crystal properties. Karlheinz Schwarz, Techn. Universitat, Wien, ISBN 3-9501031-1-2
Ceperley DM, Ader BI (1980) Phys Rev Lett 45:566–569; parametrized in Perdew J P, Zunger A Phys Rev B 8:4822–4832
Ceperley DM, Ader BI (1980) Phys Rev Lett 45:566
Perdew JP, Burke S, Ernzerhof M (1996) Phys Rev Lett 77:3865
Engel E, Vosko SH (1993) Phys Rev B 47:13164
Tran F, Blaha P (2009) Phys Rev Lett 102:226401
Reshak AH, Stys D, Auluck S, Kamarudin H (2011) Mater Chem Phys 130:458
Acknowledgement
This work was supported from the institutional research concept of the project CENAKVA (No. CZ.1.05/2.1.00/01.0024), the Grant No. 152/2010/Z of the Grant Agency of the University of South Bohemia. School of Material Engineering, Malaysia University of Perlis, Malaysia. SA would like to thank NPL for the J C Bose Fellowship. For I.Kityk, his work was supported by Polish National Science Centre (under Project No. 2011/01/B/ST7/06194).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reshak, A.H., Kityk, I.V., Parasyuk, O.V. et al. X-ray photoelectron spectrum, X-ray diffraction data, and electronic structure of chalcogenide quaternary sulfide Ag2In2GeS6: experiment and theory. J Mater Sci 48, 1342–1350 (2013). https://doi.org/10.1007/s10853-012-6879-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-012-6879-z