
Abstract
A nanosized carbonaceous material was derived from bacterial cellulose (BC). BC, which is produced by bacteria as nanosized material, possesses high degree of crystallinity of 90 %, was pyrolysed at 950 °C and physically activated with CO2 to produce a nanosized activated carbon material. The pyrolysis of BC yielded a carbonaceous material (carbon yield of between 2 and 20 %) with a relatively low D- to G-band ratio (between 2.2 and 2.8), indicating that the carbonaceous material possesses a graphitic structure. Two different BC materials were pyrolysed—a loose fibrous (freeze-dried) and dense paper form. It was observed that a carbon nanofibre-like material was produced by the pyrolysis of the loose fibrous form of BC. The electric double layer (EDL) capacitance and the area-normalised specific capacitance in K2SO4 solution were as high as 42 F g−1 and 1,617 F cm−2, respectively. The EDL capacitance was also compared to commercially available activated carbon (YP-50F).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Energy storage devices can be categorised into three categories; fuel cells, batteries and capacitors. Conventional batteries have high energy density, but low power density [1]. This implies that batteries are able to store sufficient energy for commercial use, but deliver low power. On the other hand, conventional capacitors possess high power density and hence, capacitors are able to deliver high power [2] yet store little energy. It is highly desirable to manufacture energy storage devices that combine both high energy and power densities such that they can store sufficient energy and are able to deliver high power. Therefore, numerous research efforts have been directed into the development of high power batteries [3, 4] and supercapacitors [2, 5–8]. Supercapacitors or electric double layer capacitors (EDLC) utilise the electrochemical double layer formed between a solid surface and an electrolyte solution to store energy [5]. EDLC have numerous advantages over conventional batteries and capacitors, such as a longer cycle life [7], higher energy densities [8] and rapid energy charge–discharge cycles [9].
Metal oxides [10, 11] and polymers [12, 13] have been considered as active electrode materials for EDLC. However, activated carbons remain the most frequently used electrode material for EDLC. Activated carbons are usually derived from coal, wood, coconut shells and fruit stones [14]. These carbons are of low cost, have high surface areas (~2,500 m2 g−1), are widely available and have an established electrode production technology [9]. Carbon nanotubes (CNT) are also a potential candidate as the active electrode for EDLC due to their high specific surface area and good conductivity [15–18]. More recently, the utilisation of lignocellulosic fibres as a carbon source for EDLC has been studied extensively [19–21]. Lignocellulosic fibres have attracted much attention due to their wide availability, low cost and porous nature, which usually results in high surface area of the carbon produced after pyrolysis and physical activation. Various types of lignocellulosic fibres have been studied as potential activated carbon source—wood [22], wheat straw [20], cotton stalk [23–25], jute [26], oil palm [27], coconut husk [28] and sea grass [29] fibres.
Studies have also been conducted on the carbonisation of nanosized cellulose, namely bacterial cellulose (BC) and tunicate-derived cellulose whiskers [30–32]. The synthesis of mesoporous carbon and carbon nanoneedles from cellulose nanowhiskers was reported recently [33, 34]. The study on the carbonisation of BC, however, focused on the effects of drying the cellulose from different solvents on the surface area of the carbonised cellulose. To the best of our knowledge, no studies have been performed to evaluate the physical activation of pyrolysed nanosized BC and its application for EDLC. Here, we provide a comprehensive investigation into the physical properties of physically activated carbonaceous nanomaterials derived from BC and its potential application as an active electrode material for EDLC. The capacitance of the carbon produced by the pyrolysis of BC was compared with the capacitance of commercially available activated carbon for supercapacitors (YP-50F).
Experimental
Materials
Potassium sulphate (K2SO4) solution (Fluka, 0.5 M in water) was purchased from Sigma-Aldrich. Hydrochloric acid (Alfa Aesar, 0.1 N), potassium hydroxide (Alfa Aesar, 0.1 N) and acetylene black (50 % compressed, purity >99.9 %, Alfa Aesar) were purchased from VWR. Sodium hydroxide (purum grade, pellets) was purchased from Acros Organics. Polytetrafluoroethylene (Fluon®) was purchased from AGC Chemicals. Carbon nanofibres (CNF) and a high surface area activated carbon (YP-50F) were used as benchmark for comparison and were kindly supplied by Pyrograf Products Inc (PR-19 PS, Cerdaville, Ohio, USA) and Kuraray Chemicals (Canoga Park, CA, USA), respectively. Nitrogen (purity ≥ 99.998 %) and carbon dioxide (purity ≥ 99.8 %) were purchased from BOC (Morden, UK). All the materials were used as received without further purification. BC was extracted from commercially available Nata-de-Coco (CHAOKOH coconut gel in syrup, Ampol Food Processing Ltd, Nakorn Pathom, Thailand).
Extraction and purification of BC
The extraction and purification of BC from Nata-de-Coco were reported elsewhere [35]. Briefly, the Nata-de-Coco gels from five jars of Nata-de-Coco were rinsed three times with 5 L of de-ionised water to remove the majority of the sugar syrup. After this washing step, the gels were blended for 1 min in 5 L of de-ionised water using a laboratory blender (Waring Blender LB20EG, Christison Particle Technology, Gateshead, UK) and homogenised for 2 min (Polytron PT 10-35 GT, Kinematica, Lucerne, Switzerland). The homogenised gel was centrifuged at 14,000g to remove the excess water. In order to obtain pure BC, the centrifuged BC gel was purified by re-dispersing it in 5 L of 0.1 M NaOH solution, heated to 80 °C for 20 min to remove any soluble polysaccharides and microorganisms [36]. This purified BC was then successively centrifuged and homogenised using de-ionised water to neutral pH.
Preparation of freeze-dried BC and dense BC paper
BC was prepared in two different forms before pyrolysis; as loose nanofibrils (freeze-dried) and in dense paper form. Freeze-dried BC was prepared by dispersing the previously extracted and purified BC in water at a concentration of 0.4 (g mL−1) % using a homogeniser. The dispersion was poured into a Petri dish, flash frozen in liquid nitrogen and freeze-dried (Heto PowerDry LL1500 Freeze Dryer, Thermo Scientific, UK).
Dense BC paper was manufactured following BS EN ISO 5269:2005. The previously extracted and purified BC was dispersed in water at a concentration of 0.1 (g mL−1) % using a homogeniser. This dispersion was filtered under vacuum using a filter paper (diameter: 125 mm, particle retention: 5–13 μm, VWR, Lutterworth, UK). The filter cake was wet pressed three times at a force of 410 kN. The wet-pressed filter cake was then dried at room temperature at 50 % RH to produce a dense BC paper. The loose fibrous (freeze-dried) BC and dense (paper) BC are referred to as FDBC and BCP, respectively, hereafter.
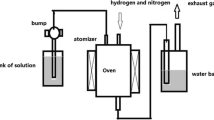
Pyrolysis and physical activation of BC
BC was pyrolysed and physically activated in a chamber furnace (ECF 12/30, 28 L chamber capacity, Lenton, Hope Valley, UK). Before pyrolysis of BC, the furnace was purged with N2 at 2 L min−1 for 15 min. BC was pyrolysed at 950 °C for 30 min under continuous N2 flow (2 L min−1) at a heating rate of 5 °C min−1. Physical activation of BC consisted of a pyrolysis step at 950 °C for 30 min (2 L min−1 of N2 flow), directly followed by an activation step in CO2 (0.5 L min−1) for 10 min. The pyrolysed FDBC and BCP were termed carbonised FDBC and carbonised BCP, respectively, whereas the physically activated pyrolysed FDBC and BCP were termed activated FDBC and activated BCP, respectively. The carbon yield or burn-off was calculated using the equations below:
where ΔY is the carbon yield and Δm is the burn-off, w i the initial mass before pyrolysis/physical activation and w f is the mass after pyrolysis/physical activation, respectively.
Characterisation of BC, carbonised BC, YP-50F and CNF
Scanning electron microscopy (SEM)
SEM was performed using a high-resolution field emission gun scanning electron microscope (LEO Gemini 1525 FEG-SEM, Oberkochen, Germany). It was used to characterise the morphology of BC before and after pyrolysis and physical activation. The accelerating voltage used during SEM was 5 kV. Before SEM, all the samples were fixed onto SEM stubs using carbon tabs and Cr coated with a sputter coater (K550 sputter coater, Emitech Ltd, Ashford, Kent, UK) for 1 min at 75 mA.
ζ-potential measurements
The ζ-potentials of neat, pyrolysed, activated pyrolysed BC and CNF were measured using the electrokinetic analyser (EKA, Anton Paar, Graz, Austria) based on the streaming potential method in a powder cell. In order to exclude any overlaying effects due to swelling (for BC) or extraction of water-soluble components from the samples, the pH dependency of ζ-potential was measured only after a time-dependent ζ-potential measurement was completed. During the ζ = f(t) measurement, a sample mass of approximately 50 mg was equilibrated in 1 mM KCl electrolyte solution by means of a single long time streaming ζ-potential measurement at 20 °C. The pH dependency of the ζ-potential was then measured by changing the pH of the electrolyte solution through the titration of 0.1 N HCl or KOH into KCl solution, using a titration unit (RTU, Anton Paar, Graz, Austria).
Specific surface area (BET) measurements
Nitrogen adsorption/desorption isotherms were performed to determine the specific surface area of neat, pyrolysed, activated pyrolysed BC, YP-50F and CNFs using a surface area and porosity analyser (TriStar 3000, Micrometrics Ltd, Dunstable, UK). The specific surface area was calculated by the Brunauer-Emmett-Teller (BET) equation. Before the measurement, the samples were degassed at 80 °C overnight to remove adsorbed water molecules.
Density measurements
The densities of neat, pyrolysed, activated pyrolysed BC, YP-50F and CNFs were measured by He pycnometry (Accupyc 1330, Micrometrics Ltd, Dunstable, UK). The samples were weighed (A&D GH-252, resolution = 0.01 mg, Milton Keynes, UK) before placing them into the measuring chamber of the pycnometer. As the pressure of He rises above atmospheric value, it was expanded through a valve and this expanded volume was measured. Owing to the expansion of He, the pressure inside the chamber will decrease to a constant value. By assuming that He obeys the ideal gas law, the density of the sample was then calculated by the equation:
where m is the sample mass, V c is the chamber volume, V E is the expanded helium volume, P 1 and P 2 are the chamber’s elevated pressure and constant pressure, respectively.
Thermal gravimetric analysis (TGA)
The thermal degradation behaviour of neat, pyrolysed, activated pyrolysed BC, YP-50F and CNFs was characterised by TGA (TGA Q500, TA Instruments, UK). Samples of 5 mg were heated from 20 to 700 °C in air at a heating rate and air flowrate of 10 °C min−1 and 60 mL min−1, respectively.
Raman spectroscopy
Raman spectroscopy was performed using a dispersive Renishaw 1000 confocal microscope (Reinshaw Plc, Hillesley, Wotton-under-Edge, UK). An argon ion laser (λ = 514.5 nm), which has a maximum output power of 20 mW and a spot size of 1 μm, was used for this characterisation. The laser was focussed onto the sample using a 50× objective at ambient condition and the spectra were measured with a collection time of 60 s and ten co-additions at 10 % of the laser power from 2,000 to 1,000 cm−1. Each sample was characterised at three randomly selected locations. The Raman spectra were peak fitted using a mixed Gaussian–Lorentzian (G/L ratio of 30:70) curve to extract the structural parameter. D- to G-band ratio (ID/IG) was calculated by taking the integrated area under the fitted curves corresponding to the D-band (1,350 cm−1) and G-band (1,590 cm−1), respectively. The in-plane graphitic crystallite size [37] L a was calculated from the D- to G-band intensity ratio by Eq. 3. This equation is valid for a Raman laser with (λ = 514.5 nm) [38].
Determination of the electric double layer capacitance
Cyclic voltammetry (CV) was performed to characterise the double layer capacitance of the produced carbonaceous materials. It was conducted using an electrochemical measurement system (Solartron 1287, Solartron Analytical, Farnborough, UK) with a three-electrode cell. Pt wire and Ag/AgCl electrodes were used as the counter and reference electrodes, respectively. In order to fabricate the working electrode, the samples were dispersed along with acetylene black and polytetrafluoroethylene (PTFE) at a ratio of 87:10:3 wt% in ethanol using an ultrasonic bath. The slurry was then applied onto two flat nickel foams (RCM-4573, RECEMAT International BV). The foams coated with the carbon material were dried at 40 °C overnight to remove any residual ethanol, then sandwiched and compressed under a weight of 250 kg for 5 min. Acetylene black was used to reduce the Ohmic resistivity of the active electrode material [39] and PTFE was used as binder. It has been shown that acetylene black does not affect the capacitance of the active electrode material [40]. The double layer capacitance was measured in a 0.5 M K2SO4 electrolyte solution. CV profiles were obtained between −0.2 and 0.2 V at various scan rates (1, 5, 10 and 20 mV s−1). The capacitance was calculated using
where C, I a, I c, m and \( \frac{{{\text {d}}E}}{{{\text {d}}t}} \) are the specific double layer capacitance, the current corresponds to the anodic and cathodic part of the CV curves, mass of the active samples and the scan rates, respectively.
Results and discussion
BET surface area of neat BC and BC-derived carbon
The properties of both BC starting materials, i.e. dense paper and loose freeze-dried (see Fig. 1), and the carbon samples produced from BC are given in Table 1. It should be noted that the table does not contain the properties of carbonised and activated freeze-dried BC (FDBC). This is a direct result of low carbon yield (~2.3 %) and large surface area (~166 m2 g−1) of carbonised FDBC. The physical activation of the carbon using CO2 involves the burning off of carbonaceous material to produce pores with CO as a by-product [14]. As a result of the high burn-off after pyrolysis, the physical activation process will remove any residual material left over after the pyrolysis of FDBC. It is, however, possible to physically activate carbonised BC paper (BCP) as it had a higher carbon yield and smaller exposed surface area (see Table 1). It can also be seen from Table 1 that the carbon yield of BCP is substantially higher than that of FDBC, which might be a direct result of the exposed surface area.

Scanning electron micrographs of a freeze-dried BC, b carbonised freeze-dried BC, c carbonised BC paper, d activated pyrolysed BC paper, e carbon nanofibres and f activated carbon YP-50F. The scale bar in figures a, b, e and f represent 200 nm whereas the scale bar in figures c and d represent 1 μm
The total surface area of the samples (carbonised FDBC, carbonised BCP and activated pyrolysed BCP) increased when it was pyrolysed/carbonised and physically activated (see Table 1). The BET surface area of our carbon materials is in agreement with the surface area of activated carbon produced by pyrolysis and activation of lignin and lignocellulosic material (sea grass), respectively [29]. The pyrolysis and activation process increased the pore area more significantly compared to the external surface area for all carbon samples (Table 1). The diffusion of CO2 into the sample removes amorphous carbon and created a porous structure within the sample through the formation of CO [14]. It is well known that the pyrolysis process will result in micropore and mesopore formation [26], whereas the activation process will widen existing pores [41].
Morphology of carbonised BC
The SEM images of the samples produced from BC are shown in Fig. 1. FDBC possesses a fibrous structure with a diameter of approximately 50 nm and several micrometres in length (Fig. 1a). When FDBC is pyrolysed, the carbon retains the fibrous structure of the BC precursor (Fig. 1b). The pyrolysis of FDBC produced a carbon nanofibre-like material. Figure 1c, d show the morphology of pyrolysed BCP and activated pyrolysed BCP, respectively. There is no observable porosity in the pyrolysed BCP. The physical activation of carbonised BC paper, on the other hand, produced a porous structure within the sample.
pH-dependent streaming ζ-potential behaviour of BC and BC-derived carbon
ζ-potential provides information regarding the surface chemistry of a material and the formation and composition of the double layer when this material is in contact with an electrolyte solution. The ζ-potential is generally assumed to approximate the electrochemical potential at the boundary between the immobile (Stern) layer and the diffusive (Gouy) part of the double layer [42]. Figure 2 shows the streaming ζ-potential of BC and carbonised BC samples in a KCl electrolyte as a function of pH. The formation of electrochemical double layer is predominantly due to the adsorption of electrolyte ions onto the surface and the dissociation of Brønsted acid/base groups. Generally, ζ-potential shows a plateau at high pH for acidic surfaces or a plateau at low pH for basic surfaces as all dissociable functional groups are either fully deprotonated for the case of acidic surfaces (COO−) or fully protonated for the case of basic surfaces and basic oxides. In this study, a negative ζplateau can be seen for BC and cellulose-derived carbonaceous material (with the exception of CNF). This is an indication that the surface of the substrate has an acidic surface character due to the deprotonation of functional groups occurring at high pH. As the pH decreased, the ζ-potential becomes more positive due to (i) protonation of functional groups and (ii) adsorption of protons (H3O+). CNFs on the other hand, possess a basic surface (Fig. 2), as indicated by a positive ζplateau. This is consistent with the ζ-potentials measured for oxidised carbon fibres [43]. At pH between 5 and 9, the surface is positively charged (positive ζ-potential value). This is indicative that the surface contains functional groups, such as basic oxides [44]. The ζ-potential increased drastically as pH decreased from 5.5 to 3 due to the adsorption of protons.

pH dependent ζ-potential of neat BC and carbon samples derived from BC
Table 2 tabulates the isoelectric points iep, the point at which no net charge is present on the surface (ζ = 0) and the ζ-potential plateau values. The iep of BCP is higher than that of FDBC due to the fact that more hydroxyl groups are involved in the hydrogen bonding of the cellulose to form a dense paper. This resulted in a reduced amount of exposed hydroxyl groups and therefore a higher iep compared to FDBC. It also can be seen that the iep of BC (FDBC and BCP) shifted to higher pH upon carbonisation and physical activation. The shift of the iep to higher pH is a result of the decomposition of dissociable functional groups due to the pyrolysis of BC. Ishimaru et al. [45] have studied the carbonisation behaviour of cotton cellulose. It was found that dehydration, decarbonylation and decarboxylation on cellulose are the major reactions that occur between 800 and 1,000 °C. The hydroxyl groups on BC are decomposed during pyrolysis, leading to an increase in the iep towards higher pH. In addition to this, the high oxygen to carbon ratio of BC will lead to the development of oxygen-containing functional groups and crosslinking of polyaromatic stacks after pyrolysis [45–47]. The crosslinking of the polyaromatic stacks led to a more hydrophobic surface. Both of these factors led to the observed decrease of ζplateau after pyrolysis. This is because water molecules do not like to adsorb on the hydrophobic carbon surface on which now more electrolyte ions adsorb.
It can also be seen that the magnitude of the ζplateau increased from −12.5 to −5 mV. The physical activation increased the hydrophilic character of activated carbon, as indicated by the increase in the magnitude of ζplateau. These shifts in iep and ζplateau can be attributed to the changes in chemical structure of the surface and the decrease in acidic functional groups as a result of physical activation. This result is consistent with the findings obtained by Julien et al. [48], who found that the magnitude of ζplateau increased with increasing activation temperature due to the burn-off of functional groups from activated carbon.
Thermal behaviour of BC and carbon samples derived from BC
Figure 3 shows the thermal degradation behaviour of neat BC and carbon samples produced from BC in air. Their onset thermal degradation temperatures are tabulated in Table 2. Two distinct weight loss steps for both BC forms—FDBC and BCP—can be observed, indicating two different types of thermal decompositions. The initial weight loss between 300 and 350 °C can be attributed to the decomposition of smaller molecular fragments on BC and the second decomposition (350–500 °C) is attributed to the degradation of the six-member cyclic structure of cellulose (pyran) [49, 50]. Different thermal degradation behaviour can be seen for FDBC and BCP. Um et al. [51] studied the degradation behaviour of silk fibres and found that the thermal degradation behaviour of the material depends on its crystallinity, molecular weight and molecular orientation of the cellulose. Since the crystallinity and the molecular weight of both FDBC and BCP are the same, the difference in the degradation behaviour must be attributed to the difference in morphology between FDBC (loose nanofibrils) and BCP (interwoven dense structure). Different thermal degradation behaviour was also observed between carbonised FDBC and carbonised BCP. This can be attributed to the differences between the starting structures of FDBC and BCP. The pyrolysis process involves the removal of non-carbonaceous (hydrogen and oxygen) material, leaving behind the skeletal structure of polyaromatic carbon [14, 52]. Owing to the loose fibrous form of FDBC, the carbonisation of FDBC might have resulted in the possible formation of more defective carbon compared to the carbonisation of BCP, which produced more graphitic carbon. In addition to this, the fabrication of BCP stretched the cellulose nanofibrils and this stretching of nanofibrils will result in improved orientation of graphite crystallites [53].

Thermal degradation behaviour of neat BC and carbon samples derived from BC in air
Raman spectra of BC-derived carbon
Figure 4 shows the Raman spectra of BC derived carbons, in the range of 1,000 and 2,000 cm−1 wavenumbers. Two typical peaks for carbon centred around 1,350 and 1,590 cm−1 can be observed and these peaks can be attributed to the disorganised carbon (D-band) and graphitic carbon (G-band), respectively with an sp 2 configuration [54]. The fitted Raman spectra are shown as the dotted lines in Fig. 4. Table 2 shows the D- to G-band ratio (calculated using the integrated area of the fitted curves) and the in-plane graphitic crystallite size of the carbon crystallites. It can be seen that the carbon produced from BC possesses D- to G-band ratios comparable to commercially available CNF and the carbonised FDBC possessed more defective carbon (I D/I G = 2.74) compared to the carbon derived from BCP (I D/I G = 2.40). This may be due to its loose fibrous form, which possibly resulted in the formation of more defective carbon during the carbonisation process. Comparing carbonised BCP and carbonised and physically activated BCP, the physical activation process resulted in more graphitic carbon. This is not surprising, as the activation process removes the defective carbon first [14]. The shift in iep to higher pH and the decreasing I D/I G ratio indicates that the formation of graphitic carbon resulted in the loss of dissociable functional groups.

Raman spectra of carbon samples derived from BC. a Carbonised FDBC, b carbonised BC paper and c activated pyrolysed BC paper
Electric double layer capacitance of (activated) BC-derived carbons, YP-50F and CNF
The double layer capacitance of carbon samples derived from BC was examined using CV over a potential range of −0.2 to 0.2 V versus an Ag/AgCl reference electrode. A typical CV profile at various scan rates is shown in Fig. 5. The well-defined rectangular shape of the CV profile is an indication that the capacitive behaviour of the sample is predominantly due to electrostatic attraction [8] and that this capacitive behaviour is reversible [19]. It can be observed from Fig. 5 that the CV profile is stable within the chosen potential window as no oxidation and reduction peaks are observed. The capacitance of carbon samples derived from BC and CNF at various scan rates is tabulated in Table 3. Carbonised BCP has the lowest capacitance, followed by carbonised FDBC and activated pyrolysed BCP. This result is consistent with the total surface area of the samples; carbonised BCP had the lowest surface area, followed by carbonised FDBC and activated pyrolysed BCP. Comparing the capacitance of these results with commercially available CNF, the double layer capacitance of the nanosized carbon derived from BC is 337 times (carbonised FDBC) and 520 times (activated pyrolysed BCP) higher than that of commercially available CNF. This result is also consistent with the total surface area of the nanosized carbon-derived BC (see Table 1).

Cyclic voltammetry of activated pyrolysed BC paper at different scan rates
It can be seen from Table 3 that the capacitance values of carbon derived from BC are still lower than that of commercially available high performance activated carbon (YP-50F). This might be due to the high surface area of activated carbon, measured to be approximately 1,938 m2 g−1 (Table 1). However, when comparing the surface area-normalised specific capacitance of all samples (specific capacitance divided by specific surface area of the sample), carbonised FDBC and activated pyrolysed BCP showed better performance compared to YP-50F. An area-normalised specific capacitance of 1,617 and 659 F cm−2 were achieved for carbonised FDBC and activated pyrolysed BCP, respectively, compared to YP-50F of only 365 F cm−2. In addition to surface area, the double layer capacitance is also related to the thickness of the Helmholtz layer [9]. As a first approximation, the double layer capacitance is proportional to the surface area and inversely proportional to the thickness of the Helmholtz layer (Eq. 6):
where C is the capacitance of the sample, ε 0, ε r , S and d are the static permittivity of vacuum, static permittivity of the medium, surface area and thickness of the Helmholtz layer, respectively. It can also be seen that the capacitance of all the samples is dependent on the scanning rates during the CV measurements. For example, at a scan rate of 10 mV s−1, the capacitance of activated and pyrolysed BCP decreased by as much as 30 % when compared to its capacitance at 1 mV s−1. Similar reduction was also observed for YP-50F. This is a direct result of the porous nature of all the carbon samples (see Table 1 for pore area of all the carbonaceous samples). When a higher scan rate is used, the carbonaceous samples behave like a small impedance element with the current will flow predominantly along the external surface and almost no current will flow down the pores [9], Table 4.
Conclusions
A nanocarbon material was produced from BC through pyrolysis and physical activation of BC. It was found that the starting morphology of BC, either as loose nanofibrils (freeze-dried) or dense paper, affects the properties of the BC-derived carbon. When BC in its loose fibrous (freeze-dried) form was pyrolysed, a carbon nanofibre-like material was formed. However, it was not possible to physically activate this material due to its low carbon yield after pyrolysis. Dense BC paper, on the other hand, could be pyrolysed and physically activated by CO2. The total surface area of the BC derived carbon increased after physical activation as determined by BET surface area measurements. The D- to G-band ratio determined by Raman spectroscopy was found to be in the range of 2.2–2.8, which is comparable to commercially available CNFs of 2.2. The electric double layer capacitance of the activated carbon derived from BC was found to be as high as 42 F g−1 (carbonised and activated BCP) in K2SO4 electrolyte solution. This can be attributed to the high specific surface area of the materials. This high capacitance performance in electrolyte solution is much higher than the capacitance of commercially available CNF (0.17 F g−1) but still lower that that of commercially available activated carbon (71 F g−1). However, when comparing the area-normalised specific capacitance, carbonised FDBC and activated pyrolysed BCP stood out, showing a value of 1,617 and 659 F cm−2, respectively, compared to activated carbon of only 365 F cm−2. The good double layer capacitance performance shown by this nanocarbon material provides a potential new active material for supercapacitors and capacitive deionisation technology.
References
Staiti P, Minutoli M, Lufrano F (2002) Electrochim Acta 47:2795
Shi H (1996) Electrochim Acta 41:1633
Taniguchi A, Fujioka N, Ikoma M, Ohta A (2001) J Power Sources 100:117
Scrosati B, Garche J (2010) J Power Sources 195:2419
Jayalakshmi M, Balasubramanian K (2008) Int J Electrochem Sci 3:1196
Lee J, Yoon S, Hyeon T, Oh SM, Kim KB (1999) Chem Commun 21:2177
Lee SI, Mitani S, Park CW, Yoon SH, Korai Y, Mochida I (2005) J Power Sources 139:379
Xu B, Wu F, Su YF, Cao GP, Chen S, Zhou ZM, Yang YS (2008) Electrochim Acta 53:7730
Kotz R, Carlen M (2000) Electrochim Acta 45:2483
Kotz R, Gobrecht J, Stucki S, Pixley R (1986) Electrochim Acta 31:169
Ardizzone S, Fregonara G, Trasatti S (1990) Electrochim Acta 35:263
Naoi K, Suematsu S (1998) Denki Kagaku 66:896
Arbizzani C, Mastragostino M, Meneghello L (1996) Electrochim Acta 41:21
Rodriguez-Reinoso F, Molina-Sabio M (1992) Carbon 30:1111
Frackowiak E, Beguin F (2001) Carbon 39:937
Honda Y, Ono T, Takeshige M, Morihara N, Shiozaki H, Kitamura T, Yoshikawa K, Morita M, Yamagata M, Ishikawa M (2009) Electrochem Solid State Lett 12:A45
Mukhopadhyay I, Suzuki Y, Kawashita T, Yoshida Y, Kawasaki S (2010) J Nanosci Nanotechnol 10:4089
Yamada Y, Kimizuka O, Tanaike O, Machida K, Suematsu S, Tamamitsu K, Saeki S, Hatori H (2009) Electrochem Solid State Lett 12:K14
Kalpana D, Cho SH, Lee SB, Lee YS, Misra R, Renganathan NG (2009) J Power Sources 190:587
Li X, Han C, Chen X, Shi C (2010) Microporous Mesoporous Mater 131:303
Liu W, Soneda Y, Kodama M, Yamashita J, Hatori H (2007) Carbon 45:2759
Klijanienko A, Lorenc-Grabowska E, Gryglewicz G (2008) Bioresour Technol 99:7208
El-Hendawy ANA, Alexander AJ, Andrews RJ, Forrest G (2008) J Anal Appl Pyrolysis 82:272
Girgis BS, Smith E, Louis MM, El-Hendawy ANA (2009) J Anal Appl Pyrol 86:180
Deng H, Yang L, Tao GH, Dai JL (2009) J Hazard Mater 166:1514
Phan NH, Rio S, Faur C, Le Coq L, Le Cloirec P, Nguyen TH (2006) Carbon 44:2569
Tan IAW, Hameed BH, Ahmad AL (2007) Chem Eng J 127:111
Tan IAW, Ahmad AL, Hameed BH (2008) J Hazard Mater 153:709
Ncibi MC, Jeanne-Rose V, Mahjoub B, Jean-Marius C, Lambert J, Ehrhardt JJ, Bercion Y, Seffen M, Gaspard S (2009) J Hazard Mater 165:240
Ishida O, Kim DY, Kuga S, Nishiyama Y, Brown RM (2004) Cellulose 11:475
Kim DY, Nishiyama Y, Wada M, Kuga S (2001) Carbon 39:1051
Kuga S, Kim DY, Nishiyama Y, Brown RM (2002) Mol Cryst Liq Cryst 387:237
Shopsowitz KE, Hamad WY, MacLachlan MJ (2011) Angew Chem Int Ed 50:10991. doi:10.1002/anie.201105479
Silva R, Al-Sharab J, Asefa T (2012) Angew Chem Int Ed. doi:10.1002/anie.201201742
Lee KY, Blaker JJ, Bismarck A (2009) Compos Sci Technol 69:2724
Toyosaki H, Naritomi T, Seto A, Matsuoka M, Tsuchida T, Yoshinaga F (1995) Biosci Biotechnol Biochem 59:1498
Knight DS, White WB (1989) J Mater Res 4:385
Baldan MR, Almeida EC, Azevedo AF, Goncalves ES, Rezende MC, Ferreira NG (2007) Appl Surf Sci 254:600
Tashima D, Taniguchi M, Fujikawa D, Kijima T, Otsubo M (2009) Mater Chem Phys 115:69
Show Y, Imaizumi K (2006) Diam Relat Mat 15:2086
Huidobro A, Pastor AC, Rodriguez-Reinoso F (2001) Carbon 39:389
Hunter RJ (1993) Introduction to modern colloid science. Oxford University Press Inc., New York
Bismarck A, Wuertz C, Springer J (1999) Carbon 37:1019
Fuente E, Menendez JA, Suarez D, Montes-Moran MA (2003) Langmuir 19:3505
Ishimaru K, Hata T, Bronsveld P, Meier D, Imamura Y (2007) J Mater Sci 42:122. doi:10.1007/s10853-006-1042-3
Oberlin A, Villey M, Combaz A (1980) Carbon 18:347
Villey M, Oberlin A, Combaz A (1979) Carbon 17:77
Julien F, Baudu M, Mazet M (1998) Water Res 32:3414
Seifert M, Hesse S, Kabrelian V, Klemm D (2004) J Polym Sci Polym Chem 42:463
Cheng K-C, Catchmark JM, Demirci A (2009) J Bio Eng 3:12. doi:10.1186/1754-1611-3-12
Um IC, Ki CS, Kweon HY, Lee KG, Ihm DW, Park YH (2004) Int J Biol Macromol 34:107
Plaisantin H, Pailler R, Guette A, Daude G, Petraud M, Barbe B, Birot M, Pillot JP, Olry P (2001) Compos Sci Technol 61:2063
Morgan P (2005) Carbon fibers and their composites. CRC Press-Taylor & Francis Group, Boca Raton
Wang Y, Serrano S, Santiago-Aviles JJ (2003) Synth Met 138:423
Acknowledgements
The authors would like to thank the UK Engineering and Physical Research Council (EPSRC) for funding KYL (EP/F028946/1) and the Challenging Engineering programme of the EPSRC for funding JJB (EP/E007538/1). The authors would like to thank Prof. Marc Anderson from the University of Wisconsin, Madison USA for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, KY., Qian, H., Tay, F.H. et al. Bacterial cellulose as source for activated nanosized carbon for electric double layer capacitors. J Mater Sci 48, 367–376 (2013). https://doi.org/10.1007/s10853-012-6754-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-012-6754-y