
Abstract
Two new donor–acceptor (D–A) conjugated polymers P1 and P2 containing 3,4-didodecyloxythiophene and 1,3,4-oxadiazole units are synthesized via Wittig reaction methodology. Cyclic voltammetry studies reveal that the polymers are both p and n dopable, and possess low-lying LUMO energy levels (−3.34 eV for P1 and −3.46 eV for P2) and high-lying HOMO energy levels (−5.34 eV for P1 and −5.27 eV for P2). The optical band gap of the polymers is in the range of 2.25–2.29 eV, calculated from the onset absorption edge. The polymers emit orange to yellow light in the film state when irradiated with a UV light. The synthesized polymers are used to prepare polymer nanocomposites with different wt% of silver nanoparticles. The polymer nanocomposites are characterized by UV–Vis absorption spectroscopy, field emission scanning electron microscopy, and thermogravimetric analysis. Both polymers and polymer/Ag nanocomposites show good thermal stability with onset decomposition temperature around 300 °C under nitrogen atmosphere. The nonlinear optical properties of polymers and polymer/Ag nanocomposites are measured by Z-scan technique. Both polymers and polymer nanocomposites show a good optical limiting behavior. Nearly five times enhancement in the nonlinear optical properties is observed for polymer/Ag nanocomposites. The value of effective two-photon absorption coefficient (β) is in the order of 10−10–10−11 m/W. These results indicate that the synthesized polymers (P1 and P2) and their Ag nanocomposites are expected to be good candidates for application in photonic devices.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Great effort has been made over the last three decades to study and synthesize conjugated polymers [1] because of their promising applications in variety of areas such as polymer light-emitting diodes (PLEDs) [2], chemical sensors [3], rechargeable batteries [4], organic thin film transistors [5], photovoltaic cells [6], and nonlinear optics [7]. In this direction, various polythiophene based conjugated polymers have been investigated extensively because of their interesting semiconducting, electronic, and optical properties; chemical stability; and their readiness of functionalization. From the application point of view, the donor–acceptor (D–A) conjugated polymers are promising because intermolecular charge transfer interaction between the electron donor (D) and electron acceptor (A) units within the polymer chain can facilitate ready manipulation of their electronic structures (HOMO/LUMO levels) and thus electronic and optoelectronic properties [8, 9]. Further, a conjugated polymer with an alternating sequence of the appropriate donor and acceptor units in the main chain shows a decreased band gap. The presence of strong electron donor and electron acceptor units in the polymer chain increases the effective π electron delocalization. Moreover, the strong delocalization of π-electrons in the polymeric backbone determines a very high molecular polarizability and thus remarkable third-order optical nonlinearities [10, 11]. Thus, D–A type polymers are being considered as promising class of third-order nonlinear optical (NLO) materials. In this regard, several such D–A polymers are synthesized and their third-order NLO properties were investigated [12–14]. In addition, metal and semiconductor nanoparticles are also emerging as a promising class of NLO materials for nanophotonic applications [15, 16]. For instance, a large NLO response with a third-order NLO susceptibility (χ(3)) value as high as 2 × 10−5 esu has been observed for nanoporous layers of TiO2 [15]. It is advantageous to embed metal/semiconductor nanoparticles in thin polymer films for application purposes because the polymer matrix serves as a medium to assemble the nanoparticles and stabilize them against aggregation [17]. Furthermore, the nanocomposite structures are known to substantially enhance the optical nonlinearities [18]. Therefore, the third-order NLO properties of several metal/semiconductor-polymer nanocomposites have been investigated. Recently, conjugated polymers are being used as host matrix for dispersing metal/semiconductor nanoparticles [19, 20]. Such a polymer/nanoparticle composite is shown to possess a large third-order nonlinear susceptibility of the order of 10−7 esu with an ultrafast response time of 1.2 ps [21]. The nanocomposites made of Ag nanoparticles dispersed in poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] matrix have exhibited a large third-order nonlinear susceptibility of the order of 10−6 esu [22]. Higher χ(3) value has been observed for polydiacetylene–Ag nanocomposite film when compared with the pure polydiacetylene film [23].
In light of these previous works, herein we report the synthesis and characterization of two new donor–acceptor conjugated polymers (P1 and P2) containing 3,4-didodecyloxythiophene moiety as electron donor and 1,3,4-oxadiazole moiety as electron acceptor unit along with thienylvinylene linkages. In these polymers, two alkoxy substituted thiophene rings are separated by a spacer unit, 2-(ethenylphenyl)-1,3-4-oxadiazole moiety, which serve not only to planarize the polymer backbone by overcoming torsional interactions between the rings thus extending effective conjugation length, but also to increase the π-electron delocalization along the polymer backbone. Hence these polymers are expected to show narrow band gap and also to show enhanced NLO properties. The chemical structure of the repeating unit in P1 and P2 is almost similar, except that one of the 3,4-dialkoxythiophene rings in P1 is replaced by an unsubstituted thiophene ring in P2. Such a simple structural modification in the polymer chain allows studying the effect of alkoxy pendant groups on optical and electrochemical properties of the conjugated polymers. The polymers are used to prepare polymer/Ag nanocomposites with different wt% silver nanoparticles. The NLO properties of the polymers and polymer/Ag nanocomposites are investigated by Z-scan technique.
Experimental
Materials and instruments
3,4-Didodecyloxythiophene-2,5-carbonyldihydrazide (1) and diethyl 3,4-didodecyloxythiophene-2,5-dicarboxylate (5) were prepared according to the previously reported procedure [24]. Thiophene-2,5-dialdehyde was purchased from Sigma Aldrich company. Tetrabutylammonium perchlorate (TBAPC) was purchased from Lancaster (UK). All solvents and other reagents were purchased commercially and used without further purification. 1H NMR spectra were recorded with a BRUKER 400 MHz NMR spectrometer using TMS as internal reference. Elemental analyses were performed on a Flash EA 1112 CHNS analyzer (Thermo Electron Corporation). Infrared spectra of all the compounds were recorded on a NICOLET AVATAR 330 FTIR (Thermo Electron Corporation). UV–Vis absorption spectra were measured using a CINTRA-101 (GBC scientific equipment) spectrophotometer. Fluorescence emission spectra were recorded using a JASCO FP6200 spectrofluorometer. The electrochemical studies of the polymers were carried out using an AUTOLAB PGSTAT 30 electrochemical analyzer. Cyclic voltammograms were recorded using a three-electrode cell system, with a glass carbon disk as working electrode, a Pt wire as counter electrode, and an Ag/AgCl electrode as the reference electrode. Gel permeation chromatography (GPC) was employed to obtain the molecular weight of the polymers using a Waters make GPC instrument with reference to polystyrene standards with THF as eluent. SEM images are obtained with a CARL ZEISS SUPRA 40 VP (NTS GmbH Germany) field emission scanning electron microscopy (FESEM) which was operated at 10 kV. Thermogravimetric analysis (TGA) was carried out using an EXSTAR TG/DTA 7000 (SII Nanotechnology Inc.) thermal analyzer. The X-ray diffraction pattern (XRD) was recorded on a JEOL X-ray diffractometer using Cu-Kα radiation operated at 30 kV and 20 mA.
Synthesis of monomers and polymers
The synthetic route for the synthesis of monomers and polymers is shown in Scheme 1. The detailed synthetic procedure is as follows.

Synthetic route of monomers and the polymers
Synthesis of 3,4-bis(dodecyloxy)-N′2,N′5-bis(4-methylbenzoyl)thiophene-2,5-dicarbohydrazide (2)
To a mixture of dihydrazide 1 (1 g, 1.75 mmol) and pyridine (0.1 mL) in NMP (10 mL), 4-methyl benzoyl chloride (0.596 g, 3.86 mmol) was added slowly at room temperature. The reaction mixture was stirred at room temperature for 12 h. After completion of the reaction (progress of the reaction was monitored by TLC), it was poured into excess of ice cold water to get a precipitate. The obtained precipitate was collected by filtration, washed with water, and dried in oven. The crude product was recrystallized from ethanol/chloroform mixture to get the pure product as white solid in 80 % yield. M.P: 98–100 °C.
1H NMR (400 MHz, CDCl3) δ: 10.23 (s, 2H, >NH), 9.19 (s, 2H, >NH), 7.75 (d, 4H, Ar), 7.27 (d, 4H, Ar), 4.30 (t, 4H, –OCH2), 2.42 (s, 6H, Ar–CH3), 1.99–1.24 (m, 40H, –(CH2)10–), 0.87 (t, 6H). FTIR √ (cm−1): 3380 and 3279 (>N–H), 2924 and 2853 (–C–H), 1597 (>C=O), 1449, 1368, 1282, 1070, 734. Element. Anal. Calcd. For C46H68N4O6S: Calcd. C 68.62, H 8.52, N 6.96, S 3.97; Found C 68.58, H 8.50, N 6.94, S 4.01.
Synthesis of 5,5′-(3,4-bis(dodecyloxy)thiophene-2,5-diyl)bis(2-p-tolyl-1,3,4-oxadiazole) (3)
A mixture of compound 2 (1 g) was refluxed with phosphorus oxychloride (10 mL) under nitrogen atmosphere for 6 h. The reaction mixture was then cooled to room temperature and poured into excess of ice cold water. The resulting precipitate was collected by filtration, washed with water, and dried in oven. The crude product was further purified by recrystallization from ethanol/chloroform mixture to get the pure product as white solid in 85 % yield. M.P: 100 °C.
1H NMR (400 MHz, CDCl3) δ: 8.01 (d, 4H, Ar), 7.34 (d, 4H, Ar), 4.31 (t, 4H, –OCH2), 2.45 (s, 6H, Ar–CH3), 1.91–1.25 (m, 40H, –(CH2)10–), 0.87 (t, 6H). FTIR √ (cm−1): 2914 and 2848 (–C–H), 1556 (C=N), 1481, 1464, 1350, 1051, 958. Element. Anal. Calcd. For C46H64N4O4S: Calcd. C 71.83, H 8.39, N 7.29, S 4.16; Found C 71.85, H 8.36, N 7.30, S 4.18.
Synthesis of 5,5′-(3,4-bis(dodecyloxy)thiophene-2,5-diyl)bis(2-(4-(bromomethyl) phenyl)-1,3,4-oxadiazole) (4)
A mixture of compound 3 (0.5 g, 0.65 mmol), N-bromosuccimide (0.25 g, 1.43 mmol), and 5 mg of benzoyl peroxide in benzene (5 mL) was refluxed for 6 h. After completion of reaction (progress of the reaction was monitored by TLC), solvent was evaporated, 20 mL of water was added, and stirred for 2 h. The resulting solid was filtered off. The crude product was recrystallized from ethyl acetate/chloroform mixture to get the pure product as yellowish solid in 60 % yield. M.P: 124–126 °C.
1H NMR (400 MHz, CDCl3) δ: 8.12 (d, 4H, Ar), 7.57 (d, 4H, Ar), 4.54 (s, 4H, Ar–CH2Br), 4.32 (t, 4H, –OCH2), 1.90–1.25 (m, 40H, –(CH2)10–), 0.87 (t, 6H). FTIR √ (cm−1): 2918 and 2850 (–C–H), 1583, 1550 (C=N), 1461, 1360, 1271, 1221, 1048, 956, 813, 725. Element. Anal. Calcd. For C46H62Br2N4O4S: Calcd. C 59.59, H 6.75, N 6.05, S 3.45; Found C 59.56, H 6.74, N 6.10, S 3.42.
Synthesis of 2,2′-(3,4-(bisdodecyloxy)thiophene-2,5-diyl)bis[5-((4-triphenylphosphonionmethyl) phenyl)-1,3,4-oxadiazole]dibromide (M1)
A mixture of compound 4 (0.5 g, 0.53 mmol) and triphenylphosphine (0.31 g, 1.18 mmol) in DMF (5 mL) was refluxed for 12 h. The reaction mixture was cooled to room temperature and poured into 25 mL of ethyl acetate. The resulting precipitate was filtered off, washed with ethyl acetate, and dried to get the product as buff solid in 90 % yield.
1H NMR (400 MHz, CDCl3) δ: 7.89–7.24 (m, 38H, Ar), 6.12–6.08 (d, 4H, Ar–CH2–), 4.30 (t, 4H, –OCH2), 1.89-1.25 (m, 40H, –(CH2)10–), 0.86 (t, 6H). FTIR √ (cm−1): 2918 and 2850 (–C–H), 1581, 1489, 1431,1371, 1106, 1048, 848, 717. Element. Anal. Calcd. For C82H92Br2N4O4P2S: Calcd. C 67.84, H 6.39, N 3.86, S 2.20; Found C 67.80, H 6.40, N 3.84, S 2.22.
Synthesis of (3,4-bis(dodecyloxy)thiophene-2,5-diyl)dimethanol (6)
To a solution of diethyl 3,4-didodecyloxythiophene-2,5-dicarboxylate 5 (1 g, 1.67 mmol) in diethyl ether (10 mL), lithium aluminum hydride (LiAlH4) (0.15 g, 4.18 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 1 h. After completion of reaction (monitored by TLC), the resulting mixture was quenched with saturated NH4Cl solution. The residue obtained was filtered through Celite, which was washed with dichloromethane. Then, the filtrate was washed with water, dried with MgSO4, and concentrated to get the product as white solid in 80 % yield. M.P: 46–48 °C.
1H NMR (400 MHz, CDCl3) δ: 7.11 (br, 2H, –OH), 4.72 (s, 4H, –CH2OH), 4.02 (t, 4H, –OCH2), 1.80–1.26 (m, 40H, –(CH2)10–), 0.88 (t, 6H). FTIR √ (cm−1): 3311 (–OH), 2915 and 2848 (–C–H), 1500, 1462, 1427, 1364, 1240, 1086, 992. Element. Anal. Calcd. For C30H56O4S: Calcd. C 70.26, H 11.01, S 6.24; Found C 70.24, H 11.0, S 6.22.
Synthesis of 3,4-bis(dodecyloxy)thiophene-2,5-dicarbaldehyde (7)
To a solution of compound 6 (0.5 g, 0.97 mmol) in diethyl ether (5 mL), 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) (0.66 g, 2.29 mmol) was added portionwise at room temperature. The reaction mixture was stirred at room temperature for 72 h. After completion of reaction (monitored by TLC), the solvent was evaporated. The solid residue obtained was redissolved in 50 mL of n-hexane and filtered off. The organic layer was washed with water several times, dried with MgSO4, and concentrated. The obtained crude solid product was recrystallized with ethanol to get the pure product as white crystals in 60 % yield. M.P: 38 °C.
1H NMR (400 MHz, CDCl3) δ: 10.09 (s, 2H, –CHO), 4.26 (t, 4H, –OCH2), 1.83–1.26 (m, 40H, –(CH2)10–), 0.88 (t, 6H). FTIR √ (cm−1): 2914 and 2848 (–C–H), 1656 (–C=O), 1480, 1430, 1366, 1263, 1186, 1037, 798. Element. Anal. Calcd. For C30H52O4S: Calcd. C 70.82, H 10.31, S 6.29; Found C 70.84, H 10.30, S 6.28.
Synthesis of polymer, P1
A solution of sodium (23 mg, 1.032 mmol) in 3 mL of anhydrous ethanol was added dropwise at ambient temperature under argon atmosphere to a mixture of compound 7 (107 mg, 0.210 mmol) and M1 (0.3 g, 0.206 mmol) in chloroform (6 mL). The mixture was stirred at room temperature for 12 h. The reaction mixture was slowly poured into 100 mL of methanol. The precipitated polymer was filtered off. The crude polymer was redissolved in chloroform and precipitated in methanol several times. After filtration and drying under vacuum, the polymer P1 was collected as yellow powder. Yield: 65 %.
1H NMR (400 MHz, CDCl3) δ: 8.13–6.71 (m, 12H, Ar and –CH=CH–), 4.33 (t, 8H, –OCH2–), 1.90–1.27 (m, 80H, –(CH2)10–), 0.88 (t, 12H). FTIR √ (cm−1): 2918, 2851, 1547, 1460, 1388, 1270, 1041, 951, 726. Element. Anal. Calcd. For (C76H112N4O6S2) n : Calcd. C 73.50, H 9.10, N 4.51, S 5.15; Found C 73.56, H 9.08, N 4.45, S 5.24.
Synthesis of polymer, P2
A solution of sodium (14 mg, 0.618 mmol) in 2 mL of anhydrous ethanol was added dropwise at ambient temperature under argon atmosphere to a mixture of thiophene-2,5-dialdehyde 8 (28 mg, 0.206 mmol) and M1 (0.3 g, 0.206 mmol) in 6 mL of chloroform. The reaction was carried out according to the procedure similar to that followed for the synthesis of polymer P1. The polymer P2 was obtained as yellow powder. Yield: 60 %.
1H NMR (400 MHz, CDCl3) δ: 8.14–6.52 (m, 14H, Ar and –CH=CH–), 4.34 (t, 4H, –OCH2–), 1.90–1.35 (m, 40H, –(CH2)10–), 0.88 (t, 6H). FTIR √ (cm−1): 2918, 2849, 1551, 1485, 1453, 1376, 1222, 1044, 950. Element. Anal. Calcd. For (C52H66N4O4S2) n : Calcd. C 71.36, H 7.61, N 6.41, S 7.31; Found 71.28, H 7.55, N 6.48, S 7.37.
Synthesis of silver nanoparticles and polymer nanocomposites
To a stirred solution of 30 mg AgNO3 in 150 mL of distilled water, 90 mg trisodium citrate was added. After stirring for 30 min, the resulting solution was exposed to UV irradiation for photoreduction of silver ions. After centrifuging, water washing, and three cycles of ultrasonication, yellow Ag colloidal nanoparticles of about 50 nm are obtained. To prepare polymer (P1 and P2)/Ag nanocomposites, 0.5, 1, and 2 wt% silver nanoparticles were dispersed in the polymer solution. The solution was sonicated for 1 h to allow uniform distribution of nanoparticles and to prevent the dispersed nanoparticles from aggregation.
Z-scan measurements
The Z-scan technique, developed by Sheik-Bahae et al. [25] was used to measure the nonlinear absorption coefficient and nonlinear refractive index of materials. The “open aperture” Z-scan gives information about the nonlinear absorption coefficient. In the experiment, a Gaussian laser beam is used for optically exciting the sample, and its propagation direction is considered as the z-axis. The beam is focused using a convex lens and the focal point is taken as z = 0. Obviously, the beam will have maximum energy density at the focal point, which will symmetrically reduce toward either side of it, for the positive and negative values of z. In our study, the sample is placed in the beam at different positions with respect to the focus (i.e., at different values of z), and the corresponding optical transmission values are measured. Then a graph is plotted between z and the measured sample transmission (normalized to its linear transmission), which is known as the Z-scan curve. The shape of the Z-scan curve will provide information on the nature of the nonlinearity. The nonlinear absorption coefficient also can be numerically calculated from the Z-scan curve. We used a stepper-motor controlled linear translation stage in our Z-scan setup, to move the sample through the beam in precise steps. The polymer and polymer/Ag nanocomposite samples were taken in a 1 mm cuvette. The transmission of the sample at each point was measured by means of two pyroelectric energy probes (Rj7620, Laser Probe Inc.). The experiment was automated, controlled by a data acquisition program written in C++.
Results and discussion
Synthesis and characterizations of monomers, polymers, and polymer nanocomposites
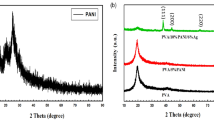
The synthetic route for the synthesis of monomers and the polymers are depicted in Scheme 1. The chemical structures of all the intermediate compounds and the polymers were confirmed by NMR spectroscopy, FTIR spectroscopy, and elemental analyses. Overall yield for all the intermediate compounds were between 60 and 85 %. The well-known Wittig reaction was performed in the presence of sodium ethoxide between the monomer M1 and the corresponding thiophene-2,5-dialdehyde in ethanol under argon atmosphere to yield the polymers, P1 and P2. To prepare monomer M1, the dihydrazide (1) was condensed with 4-methyl benzoyl chloride in N-methylpyrrolidinone (NMP) in the presence of pyridine to get 3,4-bis(dodecyloxy)-N′2,N′5-bis(4-thylbenzoyl)thiophene-2,5-dicarbohydrazide (2). Conversion of dihydrazide (1) to dicarbohydrazide (2) was confirmed by 1H NMR and FTIR spectral studies. 1H NMR spectrum of (2) showed two >NH protons as singlets at δ 10.23 and 9.19 ppm, aromatic protons as two doublets at δ 7.75 and 7.27 ppm, and tolyl methyl protons as singlet at δ 2.42 ppm. The FTIR spectrum of (2) showed sharp peaks at 3380 and 1597 cm−1 due to >NH and >C=O groups, respectively. Cyclization of compound (2) to bisoxadiazole (3) was achieved using POCl3 as the dehydrating agent. In the 1H NMR spectrum of compound (3), the disappearance of two singlet peaks due to >NH protons confirms completion of the cyclization reaction. Further, the FTIR spectrum of (3) did not show any absorption peaks corresponding to >NH and >C=O groups. Whereas a new peak appeared at 1556 cm−1 due to –C=N– stretching, indicating the formation of the oxadiazole ring. Bromination of bisoxadiazole (3) to corresponding dibromide (4) was confirmed by its 1H NMR spectrum, which showed a singlet peak at δ 4.54 ppm due to bromomethyl protons. Finally, the dibromide (4) was converted to its Wittig salt (M1) using triphenylphosphine in DMF solvent. The 1H NMR spectrum of M1 displayed complex multiple peaks at δ 7.89–7.29 ppm corresponding to the protons of triphenylphosphine and phenyl groups. The reduction of diester (5) to (3,4-bis(dodecyloxy)thiophene-2,5-diyl)dimethanol (6) was carried out using lithium aluminum hydride (LiAlH4). 1H NMR spectrum of (6) showed a broad peak at 7.11 ppm due to –OH proton and singlet peak at δ 4.72 ppm due to –CH2OH protons, whereas the FTIR spectrum displayed sharp peak at 3311 cm−1 indicating the presence of –OH groups. Conversion of bisalcohol (6) to dicarbaldehyde (7) was achieved using DDQ as the oxidizing agent. The 1H NMR spectrum of (7) showed two aldehydic (–CHO) protons as a singlet at δ 10.09 ppm whereas its FTIR spectrum exhibited a sharp peak at 1656 cm−1 due to >C=O groups. The 1H NMR spectrum of P1 displayed complex multiple peaks at δ 8.13–6.71 ppm corresponding to the aromatic and vinylic protons. A triplet peak at δ 4.33 ppm is due to the –OCH2– protons of the alkoxy chains of the thiophene ring, and the multiple peaks in the range 1.90–0.88 ppm are due to the –(CH2)10–CH3 protons of the alkoxy chains. The 1H NMR spectrum of P2 displayed a similar spectral pattern. The average molecular weights of the polymers were measured by GPC with reference to polystyrene standards. The number averaged molecular weight (M n ) of P1 and P2 are found to be 16,100 and 8,200, respectively, with molecular weight distribution (PD) 2.0 for P1 and 1.4 for P2. The polymers showed good solubility in common organic solvents such as CHCl3, THF, and chloro benzene, resulting from the alkoxy chains at 3 and 4 positions of the thiophene ring. The yellow colloidal silver nanoparticles were prepared by photoreduction method under UV irradiation using trisodium citrate as a stabilizing agent [26]. The FESEM image of the colloidal silver nanoparticles is displayed in Fig. 1a, which shows that the average size of the nanoparticles is in the range 25–50 nm. The structure of the prepared colloidal silver nanoparticles was investigated by X-ray powder diffraction (XRD) analysis. Typical XRD pattern of the silver nanoparticles is as shown in Fig. 2 which is well in agreement with the pattern of silver nanoparticles reported in the literature [27]. The prominent peaks at 2θ values of 38°, 44.4°, 64.6°, 77.6°, and 81.5° represents the (111), (200), (220), (311), and (222) Bragg’s reflections of fcc structure of silver particles. The average size of the particles is estimated to be 50 nm by Scherrer’s equations. Figure 1b shows the FESEM image of the P1/Ag nanocomposite with 1 wt % silver nanoparticles. A uniform distribution of silver nanoparticles was observed with particles sizes ranging from 20 to 50 nm.

FESEM image of a colloidal silver nanoparticles and b P1/1 wt% Ag nanocomposite film (inset: magnified image, mag. = 100 KX)

XRD spectrum of colloidal silver nanoparticles
Linear optical properties
The UV–Vis absorption spectra could provide a good deal of information on the electronic structure of conjugated polymers. Figure 3a, b shows the absorption spectra of the polymers in dilute chloroform solution, polymer thin films, yellow silver nanoparticles, and polymer/Ag nanocomposites with different concentration of silver nanoparticles. Polymer P1 in chloroform solution displayed an optical absorption maximum at 395 nm, which is attributed to the π–π* transition. The absorption spectrum of the polymer retained similar profiles regardless of the medium. This shows that P1 has no additional π-stacking while going from solution to film state. This could be due to the larger interchain distance imposed by the dodecyl chains attached to two thiophene rings which leads to a decrease in π-stacking interactions in the solid state. The optical band gap determined by the onset absorption of P1 in film state is 2.29 eV. The absorption spectrum of polymer P2 in chloroform solution displayed an optical absorption maximum at 395 nm while in the solid state the π–π* transitions showed a red shift of 21 nm with a shoulder in the shorter wavelength region at 368 nm. Obviously, the red shift in the absorption maximum of P2 in the film state is due to the π-stacking effect. Unlike in P1, the repeating unit in P2 contains pendant alkoxy groups only on one of the thiophene ring which results in lesser interchain distance in P2 as compared to that in P1. Hence π-stacking interactions could be operative in P2 in the solid state. The optical band gap of P2 determined in film state is 2.25 eV. Yellow silver colloidal nanoparticles showed an absorption peak located at about 417 nm, which results from the surface plasmon resonant oscillation of the free electrons in Ag nanoparticles. Further, the broadening in the absorption spectra was observed for nanocomposites of both the polymers (P1 and P2) with different concentration of silver nanoparticles. This occurs because the surface plasmon resonance band (SPR) in silver overlaps with the absorption region of the polymers, i.e., around 400 nm. In addition, the intensity of the absorption band increased with the increasing amount of silver nanoparticles in the nanocomposites. The absorption maximum of P1/Ag nanocomposites is slightly blue shifted in comparison with that of polymer P1 and Ag nanoparticles. The observed shift in the absorption maximum could be attributed to the agglomeration of the silver nanoparticles and/or change of the dielectric properties of the surrounding environment in the polymer matrix. These optical results give an evidence of some interaction existing between silver nanoparticles and the conjugated polymer chains. Similar trend in the absorption maximum was observed in the literature for polymer/Ag nanocomposites [19].

UV–Vis absorption spectra of a P1 in chloroform solution and in thin film, yellow silver nanoparticles, and P1/Ag nanocomposites with 0.5, 1, and 2 wt% silver nanoparticles and b P2 in chloroform solution and in thin film, yellow silver nanoparticles, and P2/Ag nanocomposites with 0.5, 1, and 2 wt% silver nanoparticles
Fluorescence emission spectra of the polymers in dilute chloroform solution and thin film state are as shown in Fig. 4. The emission maxima for P1 and P2 in solution are observed at 547 and 554 nm, respectively. The emission maxima of the polymers P1 and P2 in the film state showed bathochromic shifts of 18 and 15 nm, respectively, in comparison with those observed for their solution state. This can be attributed to the interchain or/and intrachain mobility of the excitons and excimers generated in the polymer in the solid state. The fluorescence quantum yields (ϕfl) of the polymers in chloroform solution were estimated by comparing with the standard of quinine sulfate (ca. 1 × 10−5 M solution in 0.1 M H2SO4 having a fluorescence quantum yield of 55 %) [28]. The quantum yield of the polymers is in the range of 40–42 %. The optical properties of P1 and P2 are summarized in Table 1.

Fluorescence emission spectra of P1 and P2 in chloroform solution and in thin film state
Thermal properties
TGA of the polymers and their nanocomposites with different concentration of silver nanoparticles were carried out under nitrogen atmosphere at a heating rate of 10 °C/min. As shown in Fig. 5a, polymer P1 is thermally stable up to a temperature of ~300 °C. As the temperature is increased above 300 °C, a sharp weight loss was observed, indicating the loss of alkoxy or alkyl chains and decomposition of the polymer backbone. The P1/Ag nanocomposites with 0.5 and 1 wt% silver nanoparticles did not show significant enhancement in the thermal stability. However, the polymer nanocomposite with 2 wt% silver nanoparticles showed a slight enhancement in the thermal stability as compared to the pure polymer. The polymer P2 and its nanocomposites showed a similar thermal behavior (Fig. 5b). Overall, it was observed that polymer/Ag nanocomposites start decomposing at higher temperatures than the polymer alone. A similar trend in the thermal behavior was observed for PVA/silver nanocomposites [27] reported in the literature. The improved thermal stability in polymer/Ag nanocomposites could be explained through the reduced mobility of the polymer chains in the nanocomposite due to the interaction between Ag nanoparticles with heteroatoms (S, N, and O) of the polymers. Consequently, the degradation process will be slowed in polymer/Ag nanocomposite. Further, in the nanocomposite, the percentage weight loss at higher temperature (say beyond 500 °C) is less for nanocomposite in comparison with that of the polymer because of the presence of thermally stable silver nanoparticles in the nanocomposites.

TGA graphs of a P1 and P1/Ag nanocomposites and b P2 and P2/Ag nanocomposites with a heating rate of 10 °C min−1
Electrochemical properties
Electrochemical behaviors of the polymers were investigated by cyclic voltammetry (CV). CV is widely used to estimate the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of the conjugated polymers because the onset oxidation and reduction potentials obtained from the cyclic voltammograms correspond to the HOMO and LUMO energy levels, respectively. Figure 6 shows the cyclic voltammograms of P1 and P2 thin films coated on glassy carbon (GC) disk electrode with 0.1 M TBAPC as the electrolyte at a scan rate of 50 mV/s at room temperature. A platinum wire was used as a counter electrode, and Ag/AgCl electrode was employed as the reference. All measurements were calibrated with ferrocene/ferrocenium (Fc/Fc+) standard (E FOC = 0.53 vs. Ag/AgCl). The polymers (P1 and P2) undergo both n-doping and p-doping processes. For P1, on sweeping cathodically, the onset of the n-doping process occured at a potential of −0.93 V with a reduction peak at −1.3 V. In the anodic scan, the p-doping onset occured at 1.07 V with an oxidation peak at 1.2 V. The oxidation and reduction potentials of P2 are listed in Table 2. The onset potentials of n-doping and p-doping processes were used to estimate the HOMO and LUMO energy levels of the polymers according to the equations [29]; E HOMO = −[E ox(onset) + 4.8 eV − E FOC] and E LUMO = −[E red(onset) + 4.8 eV − E FOC], respectively, where E ox(onset) and E red(onset) are the onset potentials for the oxidation and reduction processes of the polymer. Accordingly, the HOMO and LUMO energy levels of P1 are estimated to be −5.34 and −3.34 eV, respectively, and hence the electrochemical band gap is 2 eV. Similarly, the HOMO and LUMO energy levels of P2 are estimated to be −5.27 and −3.46 eV, respectively, and the electrochemical band gap is determined as 1.81 eV. The electrochemical band gap of the polymers is lower than that of the optical band gap. This difference is due to the creation of free ions in the electrochemical experiment compared with the one measured through UV experiments, which refers to a neutral state [30, 31]. The electron affinity of the polymers are lower than those of CN-PPV (−3.02 eV) and some poly(aromatic oxadiazole)s (−2.8 to −2.9 eV) [32]. From the high electron affinity value it can be expected that the polymer may show an increased electron injection ability which could be attributed to the electron withdrawing property of oxadiazole units in the polymers backbone. The HOMO energy levels of the polymers are almost similar to that of PPV (5.4 eV) indicating that the polymers have similar hole injection ability when they are used in PLEDs [33]. The polymers P1 and P2 possess high-lying HOMO and low-lying LUMO levels in comparison with those of the related D–A polymers [34]. Further, these polymers possess lower band gap as well. These could be attributed to the presence of additional electron-rich thiophene units in P1 and P2. Further, introduction of strong electron donor and electron acceptor units along the polymer chain increases the double bond character between the repeating units which raises the HOMO and lowers the LUMO energy levels of the polymer. As a result, the polymer will have high-lying HOMO and low-lying LUMO energy level and thus shows narrow band gap energy. The CV data of P1 and P2 are summarized in Table 2.

Cyclic voltammograms of a P1 and b P2 films cast on glassy carbon disk in 0.1 M tetrabutylammonium perchlorate (TBAPC)/CH3CN solution at 50 mV/s
The NLO properties of the polymers and polymer/Ag nanocomposites
The NLO properties of the polymers (P1 and P2) and polymer/Ag nanocomposites were investigated by the Z-scan technique using 5 ns laser pulses. The experiments were carried out in the “single shot” mode, allowing sufficient time between successive pulses to avoid accumulative thermal effects in the sample. The second harmonic output (532 nm) of a Q-switched Nd:YAG laser (MiniLite, Continuum) was used for excitation using 50 μJ pulse energy. The linear transmittance of all the samples studied was fixed between 50 and 60 %. The “open aperture” Z-scan gives information about the nonlinear absorption coefficient. All the samples show a strong optical limiting behavior; i.e., the transmission of the sample decreases as the input light intensity increases (intensity is a maximum at z = 0). In polymer systems under resonant excitation conditions, an optical limiting behavior can be attributed to effects such as excited state absorption (excited singlet and/or triplet absorption), two- or three-photon absorption (2PA or 3PA), and self-focusing/defocusing. Of these, 2PA, 3PA, and self-focusing/defocusing are electronic nonlinearities that require high laser intensities usually available only from pulsed picosecond or femtosecond lasers. Therefore, the cause of the observed optical limiting turns out to be excited state absorption. The net effect is then known as “effective” two-photon absorption (TPA) process. The nonlinear transmission behavior of the present samples can be modeled by defining an effective nonlinear absorption coefficient of the form,
can be considered, where α0 is the unsaturated linear absorption coefficient at the wavelength of excitation, I is the input laser intensity, and I s is the saturation intensity (intensity at which the linear absorption drops to half its original value). β is the effective TPA coefficient. For calculating the transmitted intensity for a given input intensity, the propagation equation,
was numerically solved. Here, z’ indicates the propagation distance within the sample. By determining the best-fit curves for the experimental data, the nonlinear parameters could be calculated. Figures 7 and 8 show the open aperture Z-scan curves obtained for polymer P1 and P2 and their nanocomposites with 0.5, 1, and 2 wt% of silver nanoparticles. Circles are data points, while the solid curve is a numerical fit according to Eq. 2. The value of effective TPA coefficient (β) for P1 and P1/Ag nanocomposites with 0.5 and 1 wt % silver nanoparticles were measured to be 4.4 × 10−11, 10.1 × 10−11, and 20.4 × 10−11 m/W, respectively. While for P2 and P2/Ag nanocomposites with 0.5, 1, and 2 wt% silver nanoparticles, the obtained values are 3.1 × 10−11, 3.8 × 10−11, 7.1 × 10−11, and 11.8 × 10−11 m/W, respectively. In case of polymer P1/Ag nanocomposite with 2 wt% of silver nanoparticles, scattering of light was observed during the experiment. Hence, NLO parameter for this composition was not estimated. An enhancement of nonlinearity in polymer/Ag nanocomposites are observed as compared to that of the pure polymer alone. Nearly five times enhancement in nonlinearity was observed in P1/Ag nanocomposite with 1 wt% silver nanoparticles. The observed larger nonlinearity could be a result of local field enhancement by the SPR of silver nanoparticles in the nanocomposite structure [23, 26]. This can be explained on the basis of the local field spatial variation, since the local electric field induced by the laser beam near and inside the silver nanoparticle can be much higher than the incident field through the SPR. This resonance is a direct consequence of dielectric confinement and can be interpreted in terms of a collective oscillation of the electrons in metal nanoparticles. Hence, the observed enhancement in NLO properties of polymer/Ag nanocomposites is due to the SPR of silver nanoparticles, which induces higher nonlinear absorption, in comparison with that of the polymer alone. In addition, in the case of the nanosecond region, the thermal effect [35] cannot be neglected but is considered to be a contribution to the nonlinear properties. The β values of all the samples are listed in Table 3. The values obtained in similar systems under similar excitation conditions are: 10−11 m/W for copper complexes containing 1,10-phenanthroline-5,6-dione and triphenylphosphine ligands [36], 10−10–10−12 m/W for copper (Cu) nanocomposite glasses [37], 3 × 10−11 m/W for functionalized carbon nanotubes [38], and 0.53 × 10−10 m/W for bismuth (Bi) nanorods [39]. Thus, the improved optical limiting behavior indicates the potential of these polymer nanocomposites to be used as materials for photonic devices. The observed nonlinearity in P1 and P2 could be attributed to the effective π-electron delocalization due to the presence of strong electron donor and electron acceptor units. Further, the presence of thienylvinylene linkages not only planarize the molecule but also increase the conjugation length of the repeating units, both contributing to the effective π-electron delocalization in the polymer chain and thus enhancing the NLO properties of the polymer. Hence, incorporation of strong electron donor and electron acceptor units in the polymer chain could be a promising molecular design to obtain polymeric materials with high nonlinear response. In addition, the NLO properties in these systems can be further enhanced by the incorporation of metal nanoparticles into the polymer matrix, forming nanocomposite structures.

Open aperture Z-scan curve of a P1, b P1/0.5 wt% Ag nanocomposite, and c P1/1 wt% Ag nanocomposite

Open aperture Z-scan curve of a P2, b P2/0.5 wt% Ag nanocomposite, c P2/1 wt% Ag nanocomposite, and d P2/2 wt% Ag nanocomposite
Conclusions
Two new donor–acceptor (D–A) conjugated polymers, P1 and P2 containing 3,4-didodecyloxythiophene and 1,3,4-oxadiazole units were synthesized by Wittig method. The chemical structures of the polymers were established by 1H NMR and FTIR spectroscopic methods. The presence of alkoxy pendants on thiophene rings was found to influence the optical properties of the polymers. Hence, polymer P1 with two alkoxy substituted thiophene rings showed no signs of π-stacking interactions in the solid state, which could be attributed to the larger interchain distance imposed by the alkoxy groups on two thiophene rings present in the polymer. Thus, the absorption spectrum of the polymer retained similar profiles regardless of the medium. Whereas polymer P2 with an unsubstituted thiophene ring showed a red shift in the absorption maxima on going from solution to solid state, indicating the presence of π-stacking interactions in the solid state. The electrochemical properties revealed that P1 and P2 possess high-lying HOMO and low-lying LUMO energy levels due to the presence of strong electron donating and electron accepting units in the polymers chain. The yellow Ag colloidal nanoparticles of average size ranging from 25 to 50 nm were obtained by the photoreduction of silver ions under UV irradiation. The polymer/Ag nanocomposites were prepared with different wt% silver nanoparticles by a simple dispersion method. The effect of incorporation of silver nanoparticles on thermal and optical properties of the polymers was investigated. As compared to the polymer, a broadening in the absorption spectrum was observed after the addition of silver nanoparticles into the polymer matrix, which could be due to the overlap of SPR band of Ag nanoparticles with the absorption region of the polymers. The NLO properties of the polymers and polymer/Ag nanocomposites were studied by open aperture Z-scan technique. All the samples showed a strong optical limiting behavior, and the incorporation of Ag nanoparticles significantly enhances the nonlinear absorption coefficient value of the polymer. The highest effective TPA coefficient (β) value of 2.04 × 10−10 m/W was obtained for P1/Ag nanocomposite with 1 wt% silver nanoparticles. The observed larger nonlinearity is a result of local field enhancement by the SPR of silver nanoparticles in the nanocomposite structure. Further, the results indicate that the NLO properties of polymers and their nanocomposites can be easily modulated by varying the concentration of Ag nanoparticles. Hence, the polymers (P1 and P2) and their nanocomposites are expected to be good candidates for new optical and nonlinear devices.
References
Skotheim TA, Elsenbaumer RL, Reynolds JR, Dekker M (1998) Handbook of conducting polymers, 2nd edn. Marcel Dekker, New York
Kraft A, Grimsdale C, Holmes AB (1998) Angew Chem Int Ed 37:402
Mcquade DT, Pullen AE, Swager TM (2000) Chem Rev 100:2537
Novak P, Muller K, Santhanam KSV, Haas O (1997) Chem Rev 97:207
Dimitrakopoulus CD, Mascaro DJ (2001) IBM J Res Dev 45:11
Zou Y, Guan Z, Zhang Z, Huang Y, Wang N, Lu Z, Jiang Q, Yu J, Liu Y, Pu X (2012) J Mater Sci 47:5535. doi:10.1007/s10853-012-6446-7
Bredas JL, Adant C, Tackx P, Persoons A, Pierce BM (1994) Chem Rev 94:243
Jenekhe SA, Li S (2000) Appl Phys Lett 77:2635
Sonmez G, Shen CKF, Rubin Y, Wudl F (2005) Adv Mater 17:897
Prasad PN, David JW (1991) Introduction to nonlinear optical effects in molecules and polymers. Wiley, New York
John Kiran A, Udayakumar D, Chandrasekharan K, Adhikari AV, Shashikala HD (2006) J Phys B 39:3747
Hegde PK, Adhikari AV, Manjunatha MG, Suchand Sandeep CS, Philip R (2011) Polym Int 60:112
Ramos Ortiz G, Maldonado JL, Hernández MCG, Zolotukhin MG, Fomine S, Fröhlich N, Scherf U, Galbrecht F, Preis E, Salmon M, Cárdenas J, Chávez MI (2010) Polymer 51:2351
Manjunatha MG, Adhikari AV, Hegde PK, Suchand Sandeep CS, Philip R (2009) J Mater Sci 44:6069. doi:10.1007/s10853-009-3838-4
Gayvoronsky V, Galas A, Shepelyavyy E, Dittrich Th, Timoshenko VYu, Nepijko SA, Brodyn MS, Koch F (2005) Appl Phys B 80:97
Venkatram N, Rao DN, Akundi MA (2005) Opt Express 13:867
Takele H, Greve H, Pochstein C, Zaporojtchenko V, Faupel F (2006) Nanotechnology 17:3499
Neeves AE, Meyer BH (1988) Opt Lett 13:1087
Sezer A, Gurudas U, Collins B, Mckinlay A, Bubb D (2009) Chem Phys Lett 477:164
Luo Q, Li X, Wang D, Wang Y, An J (2011) J Mater Sci 46:1646. doi:10.1007/s10853-010-4981-7
Hu XY, Jiang P, Ding CY, Yang H, Gong QH (2009) Nat Photonics 2:185
Hu X, Zhang J, Yang H, Gong Q (2009) Opt Express 17:18858
Chen X, Tao J, Zou G, Zhang Q, Wang P (2010) Appl Phys A 100:223
Udayakumar D, Adhikari AV (2006) Synth Met 156:1168
Sheik-Bahae M, Said AA, Wei TH, Hagan DJ, Van Stryland EW (1990) IEEE J Quant Electron 26:760
Chen X, Zou G, Deng Y, Zhang Q (2008) Nanotechnology 19:195703
Khanna PK, Singh N, Charan S, Subbarao VVVS, Gokhale R, Mulik UP (2005) Mater Chem Phys 93:117
Joshi HS, Jamshidi R, Tor Y (1999) Angew Chem Int Ed Engl 38:2721
De Leeuw DM, Simenon MMJ, Brown AB, Einerhand REF (1997) Synth Met 87:53
Hou J, Park MH, Zhang S, Yao Y, Chen LM, Li JH, Yang Y (2008) Macromolecules 41:6012
Ma CQ, Fonrodona M, Schikora MC, Wienk MM, Janssen RAJ, Bauerle P (2008) Adv Funct Mater 18:3323
Bradley DDC (1993) Synth Met 54:401
Cervini R, Li XC, Spencer GPC, Holmes A, Moratti SC, Friend RH (1997) Synth Met 84:359
Manjunatha MG, Adhikari AV, Hegde PK (2009) Eur Polym J 45:763
Liao HB, Xiao RF, Fu JS, Wang H, Wong KS, Wong KL (1998) Opt Lett 23:388
Rudreshaa BJ, Ramachandra Bhat B, Sampath Kumara HC, Shiva Kumara KI, Safakath K, Philip R (2011) Synth Met 161:535
Karthikeyan B, Anija M, Suchand Sandeep CS, Muhammad NTM, Philip R (2008) Opt Commun 28:2933
Nan H, Yu C, Jinrui B, Jun W, Werner JB, Jinhui Z (2009) J Phys Chem C 113:13029
Sivaramakrishnan S, Muthukumar VS, Sivasankara SS, Venkataramanaiah K, Reppert J, Rao AM, Anija M, Philip R, Kuthirummal N (2007) Appl Phys Lett 91:093104
Acknowledgements
Authors thank Prof. Reji Philip, Raman Research Institute, India for providing facility for Z-scan measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Murali, M.G., Dalimba, U. & Sridharan, K. Synthesis, characterization, and nonlinear optical properties of donor–acceptor conjugated polymers and polymer/Ag nanocomposites. J Mater Sci 47, 8022–8034 (2012). https://doi.org/10.1007/s10853-012-6692-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-012-6692-8