
Abstract
The effect of filler size and content in the thermal, mechanical, and electrical response of poly(vinylidene fluoride) (PVDF)/BaTiO3 nanocomposites has been investigated. Dielectric constant increases significantly with increasing filler content and decreasing filler size. Space charge effects at the interface between BaTiO3 and PVDF strongly influence the dielectric response. The electroactive β-phase of PVDF is nucleated by the presence of the ceramic filler, the effect being strongly dependent on filler size and independent on filler content. This filler/matrix interaction is also responsible for the variations observed in the activation energy of the thermal degradation of the polymer. Smaller particles lead to larger relative contact areas and are responsible for the main variations observed in the thermal, mechanical, and electrical properties of the composites.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polar oxides such as barium titanate (BaTiO3), lead zirconate titanate (Pb(Zr,Ti)O3), and SrBi2Ta2O9 and electroactive polymers such as poly(vinylidene fluoride) (PVDF) are among the most relevant ferroelectric materials. They have received increasing attention in recent years, both from a fundamental point of view and for the development of novel technological applications [1].
PVDF is a semicrystalline polymer in which the amorphous chains are embedded between the lamellar crystalline structures of the polymer matrix [1, 2]. PVDF presents at least four crystalline phases known as α, β, γ, and δ, depending on the processing conditions [1, 3, 4].
β-PVDF presents the highest electroactive properties and, until recently, this phase was exclusively obtained by mechanical stretching of films originally in the non-polar α-phase. This process results in oriented β-PVDF films with a small amount of α-phase material [1, 4, 5].
BaTiO3 is a ceramic material that exhibit piezoelectric and ferroelectric properties and crystallizes in a perovskite structure [6]. This material can exhibit five crystalline phases, hexagonal, cubic, tetragonal, orthorhombic, monoclinic, and rhombohedral crystal structure depending on temperature [7].
Nanocomposite materials have achieved large interest in order to study the effect of the small material structures within a given matrix, including the study the effect of the filler geometry and content in the overall composite response (i.e., the study of fibrous composite materials, laminated composite materials, composite materials with dispersed particles, and combinations of them [8, 9]). Composite materials are relevant due to the possibility to tailor materials properties for specific applications. In particular, piezoelectric composites are pursued due to their higher dielectric and electroactive properties when compared to the polymer matrix and due to their higher flexibility and easier production in large areas or tailored shapes when compared to ceramic or single crystal materials [10, 11].
Some works have been previously devoted to the study of PVDF/BaTiO3 composites and other similar polymer/filler systems. The main effect of BaTiO3 inclusion is the increase of the dielectric constant with increasing filler content [12], whereas the characteristics of the polymer matrix is just slightly affected as demonstrated by the small variation of the melting temperature of PVDF with BaTiO3 inclusion. The evolution of the dielectric constant with increasing BaTiO3 content follows the theoretical model of Yamada model [13]. Together with filler concentration, the dielectric properties of BaTiO3/PVDF composites will also depend on particle size [14] due to space charge effects at the interface between BaTiO3 and PVDF and domain configurations (single or multi-domain) of the BaTiO3 powders. The shape and orientation of the filler also influences the composite response, as indicated by composites containing highly oriented BaTiO3 whiskers as active phase and PVDF as matrix, prepared by micro-hole extrusion and orientation in the form of fibers. The experimental results showed that the ε, d 33, and P r in the BaTiO3 whisker–PVDF composite were considerably higher than those in the BaTiO3 powders–PVDF composite, while the loss factors followed the opposite trend [15]. Finally, it has been demonstrated that these interesting composites can be produced by up-salable processing methods, maintaining the properties of the ones prepared in laboratory conditions [16].
Despite of the aforementioned efforts proving the interesting characteristics of the composites for sensor and actuator applications, no systematic study on the effect of BaTiO3 ceramic grain size itself on the PVDF polymer phase and the macroscopic response of the material has been carried out, as most of the studies just focused on the effect of the ceramic volume fraction on the dielectric response of the composites. In this scope, it is also very relevant to address the variation of the mechanical properties of the composite as well as the degree of crystallinity of the polymer with increasing filler content, as these parameters will determine the overall macroscopic response and the range of applicability of the materials. In this sense, this work represents a comprehensive study on PVDF/BaTiO3 composites. The role played by the size and content of the ceramic particles and eventual variations in the phase of the polymer matrix was studied. Furthermore, the thermal degradation of the composite was also addressed.
In the present work the ceramic phase included was BaTiO3 instead of the most studied PVDF/PZT composites [17, 18]. PVDF/BaTiO3 composites samples with different BaTiO3 particle size and content were prepared by solution casting in the non-polar α-PVDF. Spectroscopic, dielectric, mechanical, and thermal techniques were applied in order to investigate the influence the filler size and content in the composites response.
Experimental details
BaTiO3 particles with an average size of 10 and 500 nm were purchased from Nanoamor (USA). PVDF (Solef 1010) was obtained from Solvay (Belgium) and N,N-dimethyformamide (DMF) from Merck (Germany). All materials were used as received.
Composites of PVDF with BaTiO3 were prepared by dispersing the ceramic powder in a solution of PVDF in DMF. A certain amount of ceramic nanoparticles (5 and 10% of mass fraction of PVDF) and 12 mL of DMF were mixed in an ultrasound bath for 6 h. Then, 3 g of PVDF were added to the mixture and placed in magnetic stirrer until complete dissolution of the polymer. Flexible films were prepared by spreading the suspension on a glass plate and placing it in an oven for 60 min at 120 °C. This period time insured the total evaporation of the solvent. The samples were heated at 220 °C for 10 min and cooled down to room temperature.
Scanning electron microscopy (Leica Cambridge apparatus at room temperature) was performed in order to evaluate the composites microstructure and the dispersion of the ceramic powder within the polymer matrix. All the specimens were coated with a conductive layer of sputtered gold.
Fourier transformed infrared spectroscopy (FTIR) spectra of the films were recorded on a Perkin-Elmer Spectrum 100 in ATR mode over a range of 650–3500 cm−1 with a resolution of 4 cm−1 and 32 scans were performed to each sample.
Differential scanning calorimetry (DSC) studies were performed using a Perkin-Elmer Diamond apparatus. During the experiment, the samples were ramped from 20 to 200 °C under a dry N2 environment at a rate of 10 °C/min.
Thermogravimetric analyses (TGA) were carried out using a Pyris 1 TGA–Perkin-Elmer under nitrogen atmosphere supplied at a constant 50 mL min−1 flow rate. The sample holders used were crucibles of ceramic with capacity of 60 μL. The samples were analyzed by using the same heating rate (10 ± 0.2 °C/min) between 50 and 900 °C. Further, experiments at different heating rates of 10 ± 0.2 to 30 ± 0.4 °C min−1 between 50 and 900 °C were also performed in order to evaluate the influence of the average ceramic size in the degradation kinetics of the PVDF/BaTiO3 composites.
Dynamical–mechanical measurements (dynamic mechanical analyses, DMA) were performed at room temperature in a DMA8000 apparatus from Perkin-Elmer using the tensile mode on rectangular samples with typical dimensions of 10 × 40 × 0.030 mm at a frequency scan from 0.01 to 10 Hz.
Dielectric measurements were performed using a Quadtech 1920 LCR precision meter. The real part of the complex permittivity (ε′) and the dielectric losses (tan δ) were obtained at room temperature in the frequency range of 500 Hz to 1 MHz with an applied voltage of 0.5 V. Circular gold electrodes of 5 mm diameter were vacuum evaporated onto both sides of each sample.
Results and discussion
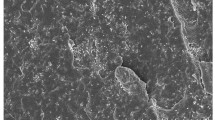
The samples were analyzed by scanning electron microscopy (SEM) in order to evaluate the dispersion of the nanofillers and the influence of the filler in the microstructure of the polymer phase [19]. Figure 1 shows a typical SEM image obtained for the PVDF/BaTiO3 composites, as the microstuctural features are similar for all filler sizes and concentrations used in this work. The obtained results show a typical spherulitic structure with small porous in the region between the spherulites which is a result of the solidification process. During the crystallization process, spherulites grow and expand absorbing the liquid polymer phase that is in the region between the crystallized polymer. At the end of crystallization, no liquid phase remains, and as a consequence a void is generated between the crystallized spherulites (Fig. 1a). The microstructure of the nanocomposite samples is very similar to the one observed for neat α-PVDF, characterized by spherulites with small concentric rings in the interior [20]. On the other hand, neat β-PVDF samples show much smaller spherulites and show a very porous structure, resulting in poor mechanical and electrical properties [5]. The backscattering images of the nanocomposites samples show the good distribution of the nanofillers (Fig. 1b).

a SEM image of the PVDF/BaTiO3 composites with 10% filler concentration and 500 nm filler size. b Corresponding backscattering image
PVDF is a semicrystalline polymer exhibiting four crystalline phases [1, 4, 5], being the β-phase the one with the highest electroactive properties. The presence of the β-phase is identified by infrared spectroscopy mainly through the characteristic absorption band at 840 cm−1 and the α-PVDF by the bands at 765, 795, 855, and 976 cm−1 [3, 21]. Figure 2 shows the infrared spectra of the pure α- and β-PVDF for composites with different contents and sizes of BaTiO3 particles. In all cases, the presence of β-phase can be observed through the presence of the absorption band at 840 cm−1. In addition, the infrared spectra also reveal an absorption band at 812 cm−1 that also indicates the presence of small contents of γ-PVDF (Fig. 2) [22].

FTIR spectra for the PVDF/BaTiO3 samples with different filler contents for average filler sizes of a 10 nm and b 500 nm
The β-phase content present in each sample was calculated from the absorption bands at 764 and 840 cm−1, characteristic of the α- and β-PVDF, respectively, with a procedure similar to the one reported in [19, 20]. Briefly, assuming that the infrared absorption follows a Lambert–Beer law, for a system containing just α- and β-phase, the relative amount of β-PVDF, F(β), can be calculated by applying Eq. 1:
where A α and A β are the absorbencies at 766 and 840 cm−1, corresponding to the α- and β-phase material and K α (6.1 × 104 cm2/mol) and K β (7.7 × 104 cm2/mol) are the absorption coefficients at the respective wave number. The calculated β-phase content for the prepared composites is presented in Table 1.
It is to notice that the γ-phase content has been neglected in this calculation due to the small amount of this phase in the composite samples, as indicated by the corresponding band strength at 812 cm−1 in the FITR spectra of Fig. 2. The error induced by this simplification will be within the 4% error estimated by the method itself [22]. It was observed that the presence of the BaTiO3 nanoparticles promotes the crystallization of the β-phase of the PVDF matrix, indicating that the ceramic filler act as a nucleating agent for this phase. Moreover, the highest amount of electroactive phase (F(β) = 82%) was achieved for the lowest concentration of ceramic particles with the smaller average size (ϕ = 10 nm) and therefore with the largest specific surface interaction area for a given filler volume fraction.
In PVDF/DMF solution, molecular interactions between the polymer and the solvent involve strong dipolar interactions between the C=O and CH2–CF2 dipole and maybe the presence of weak hydrogen bonding C=O···H–C. In this way, the dipolar and Van der Waal’s interactions that hold the polymer chains together are disrupted and the PVDF polymer is dissolved [23, 24]. These interactions also play an important role in crystallization of PVDF. Crystallization of PVDF begins with nucleation, which generally involves two steps: the formation of colloidal crystals of PVDF molecules with a critical radius within the solution and the rate-limiting organization of such clusters into crystal nuclei [25, 26].
Dipole molecules in the surface of embryonic droplets are oriented perpendicular to the interface due to the dipolar interactions [27] and PVDF nucleation could also exhibit such effects from polar solvents. In this way, dipolar interaction and hydrogen bonding at the interface between crystalline nucleus and DMF molecules lead to an all-trans planar (TTT) configuration of CH2–CF2 dipoles, when the nucleation occurs at room temperature [3, 5]. However, if a relatively large external energy, such as thermal energy is provided during the crystallization process, the forces that are at the origin of the TTT packing of the polymer chains becomes weak relatively to the thermal energy to prevent PVDF molecules to crystallize into trans-gauche conformation of the α-phase of PVDF, which is thermodynamically more stable [3, 5].
When BaTiO3 nanoparticles are added to the solution, stronger O–H···F–C hydrogen bonds are probably formed due to the high polarity of the hydroxyl groups, the ceramic nanoparticles acting in this way as nucleating agents for the PVDF crystals. The strong O–H···F–C hydrogen interaction at the BaTiO3 nanoparticles/PVDF interfaces, together with the dipolar interactions between the PVDF and DMF solvent, tend to produce locally oriented CH2–CF2 dipoles that are packed in the TTT configuration characteristic of the β-phase. Further, as demonstrated in Fig. 1, the nucleation of the β-phase is proportional to the specific area of the particles: the increase of the ceramic filler size corresponds to a decrease of the specific surface area available to promote the O–H bonding, and consequently promotes an increase of the crystallization “trans-gauche” configuration characteristic of the non-electroactive α-PVDF.
This hypothesis is further supported by a broad absorption band from 3100 to 3700 cm−1 (Fig. 3) attributed to the O–H stretch hydrogen bond [28], which does not exist for the pure polymer.

FTIR spectra for the PVDF/BaTiO3 composites and for the pure polymer (inset) in the O–H region
The abovementioned facts indicate that birth the size of the nanoparticles and their chemical structure are the key issues for the nucleation the electroactive β-PVDF phase when the composites are obtained from the melt. Previous works related to the processing of PVDF with nano and micro fillers like PZT (lead zirconate titanate) [17] or ferrite nanoparticles (CoFe2O4 or NiFe2O4) [29] reveal that there is also a critical size for the filler in order to promote the β-phase nucleation of PVDF. In the case of the PVDF/PZT composites, the smaller average size of the filler was 0.84 μm and the polymer matrix crystallizes in the α-PVDF [17, 18], but in the case of the ferrite nanoparticles (ϕ = 20–55 nm), the polymer also crystallizes from the melt mainly in the β-phase [29]. In this way there is a balance between the specific polymer/nanoparticle interphase area and therefore between the interaction energy and the thermal energy that lead to the polymer crystallization in either the β- or the α-phase.
Thermal behavior
DSC thermographs for the PVDF/BaTiO3 nanocomposites (Fig. 4) show two endothermic peaks for all samples. Multiple DSC peak structures are typical in composites and nanocomposites due to interface effects and ill crystallized parts of the sample, that melt at different temperatures than the main body of the polymer [17, 18]. In the present case, nevertheless, this behavior has a stronger contribution from the coexistence of α- and β-PVDF phases in the samples, as observed by FTIR (Fig. 1), that melt at slightly different temperatures [19, 20]. The peak at lower temperature corresponds in this way to the melting of the α-phase and the higher temperature one is related to the melting of the β-PVDF [20]. Moreover, a small peak can be observed for the samples with 10% BaTiO3 content, which is assigned to the melting of the γ-phase [22]. This phase was also identified in FTIR by the presence of the absorption band at 812 cm−1 (Fig. 2).

DSC curves of BaTiO3/PVDF composites and pure PVDF
The degree of crystallinity \( \left( {X_{\text{c}} } \right) \) of PVDF and the BaTiO3/PVDF nanocomposites was obtained by Eq. 2:
where x and y corresponds to the weight fraction of the α- and β-phase, respectively, and (ΔH 100% crystalline)α and (ΔH 100% crystalline)β are the melting enthalpies for 100% crystalline α- and β-PVDF: 93.04 and 103.4 J/g, respectively [4].
Figure 2 shows that the inclusion of BaTiO3 particles in the PVDF matrix increases the thermal stability of the composite (shift to the melting temperature to higher values), but that the degree of crystallinity decreases with increasing BaTiO3 particle content in the composite samples (Table 2). For a given filler content, the size of the filler does not influences the degree of crystallinity. The inclusion of nanoparticles causes ill crystallized regions in the interface regions contributing to the decrease in the crystallinity of the polymer [17, 18].
The interaction between the inorganic fillers and the polymer can be explored by its effect in the polymer thermal degradation, which was measured by thermogravimetric analysis (Fig. 5). The thermal degradation of the samples showed in all cases a single step degradation process, similar to the one observed in the pure polymer matrix. The onset of the degradation process shifts to higher temperatures in the polymer composites being the shift larger for the larger filler contents. This fact proves an increased stability of the polymer chains against thermal degradation that should be attributed to their interaction with the ceramic filler. On the other hand, filler size has no effect for the sizes under consideration in this work. The residual mass of the polymer observed at the end of the measurement (750 °C) is quite similar for all samples, once considered the residual mass of BaTiO3 that does not suffer thermal degradation at those temperatures.

Thermogravimetric results for a PVDF/BaTiO3 samples with different filler contents obtained at 10 °C min−1 and b for PVDF/BaTiO3 composites with 5% BaTiO3 obtained at several heating rates
The kinetics of the mass loss process can be investigated by analyzing experiments performed at different heating rates (Fig. 5). As expected, increasing heating rate shifts the onset of the degradation process to higher temperatures (Fig. 5), not affecting any of the main characteristics of the process itself. A typical model for the thermal decomposition has the following general form [30, 31]:
Here, α represents the degree of conversion of the sample under degradation, defined by:
where w 0, w t, and w ∞ are the weights of the sample before degradation, at a given time t and after complete degradation, respectively. The rate constant k(t) changes with the absolute temperature according to the Arrhenius equation and f(α) represents the net results of elementary steps, as the polymer degradation is often a chain reaction. For solid state reactions f(α) = (1 − α)n, where n is the reaction order and it is assumed to remain constant during the degradation process.
The isoconversional method of Ozawa–Flynn–Wall (OFW) [32, 33] assumes that the conversion function f(α) does not change with the variation of the heating rate for the different values of the degree of conversion α. It involves measuring the temperatures corresponding to fixed values of α from the experiments at different heating rates β. In this formalism:
where A is a pre-exponential factor, R is the gas constant (8.31 J mol−1 K−1), and E act is the activation energy of the degradation process. By plotting ln(β) versus 1/T, E act can be obtained from the slope of the resulting straight line, regardless of the reaction order of the system. The validity of this method is based on the assumption that the conversion at the peak maximum is constant for the different heating rates [32, 34]. The linear fitting for the PVDF/BaTiO3 nancomposites with 5% filler content of 10 and 500 nm filler sizes is represented in Fig. 6.

Ozawa–Flynn–Wall plots for the PVDF/BaTiO3 composites with 5% filler content: a ϕ = 10 nm and b ϕ = 500 nm filler sizes
As it was previously demonstrated for PVDF/PZT composites [35], filler size affects more than filler concentration the degradation temperature and activation energy of the polymer, the polymer degradation mechanism [36] being not significantly affected by the presence of the inorganic fillers. The present experiments for PVDF/BaTiO3 composite samples confirm this trend, verifying a strong filler size effect both for the value of the activation energy and for its behavior as a function of the conversion rate (Fig. 7).

Activation energy for PVDF and PVDF/BaTiO3 composites as a function of the conversion rate for samples with 5% filler content and different filler sizes
For the smaller conversion rates, the value of the activation energy for a given conversion rate increases for increasing filler size, as demonstrated in [35]. For the larger conversion rates the behavior is inverted due to the different trend observed for the different filler sizes and the pure polymer: whereas for the pure polymer and the smaller filler the activation energy increases with increasing conversion rate, for the larger filler size the activation energy shows a small decrease with increasing conversion rate. In this way, increasing filler size leads to a more homogeneous degradation process in terms of energy.
During the main thermal degradation of PVDF carbon–hydrogen bond scission primarily occurs which is due to the lower bond strength energy of C–H compared to C–F bond (410 and 460 kJ mol−1, respectively) [36]. The head to tail (H–T) configuration once activated by the removal of one HF molecule to form a carbon–carbon double bond, will unzip HF molecules down the polymer chain, leading to the main degradation process.
The differences on the thermal stability should be related to chemical reactions with the ceramic particles within the polymer–ceramic interfacial region or to incomplete removal of the DMA solvent due to some absorption by the ceramic particles. In this case high temperature degradation of DMA and chemical reaction with the PVDF polymer can affect its thermal degradation [37].
The observed variations on the onset temperature (Fig. 5) and the activation energy with conversion rate, points out effectively to the ceramic–polymer interface effects (interface dimension and chemical stability against temperature) that provide more stability to the polymer phase, being the effects more prominent for larger ceramic particles.
Mechanical response
The mechanical characteristics of the composite will influence the performance of the material for sensors and actuators applications. Dynamical–mechanical analysis (DMA) measurements at room temperature for all PVDF/BaTiO3 nanocomposites are presented in Fig. 8.

a Evolution of the storage modulus for PVDF and PVDF/BaTiO3 composites, b E′ at 1 Hz as a function of the filler content for the different filler sizes, and c mechanical dissipation factor evolution for PVDF and PVDF/BaTiO3 composites
The inclusion of the nanoparticles with smaller average size (ϕ = 10 nm) in the polymer matrix produces an increase of the storage modulus, which confirms that the ceramic filler works as a nucleation agent, the polymer crystallizing around the nanoparticles increasing the connectivity between the polymer chains and the filler. For the samples with higher ceramic diameter (ϕ = 500 nm) and small filler contents (5%), an increase of E′ was observed but, on the other hand, increasing filler content decreases the value of E′. These effects are related to a competition between two effects: the filler acts as nucleation agent for low filler concentrations but also decreases the connectivity of the PVDF matrix, hindering the crystal reorganization when a stress is applied. Further, due to the lower interaction strength between higher particles and the polymer matrix, some debonding and sliding of the BaTiO3 nanoparticles occur, especially for the sample with 10% ceramic filler and ϕ = 500 nm, leading to smaller E′. As already observed in other PVDF/filler systems [14, 17, 18], for higher particle average size and concentration, the fillers act as imperfections and defects in the polymeric matrix and reduce the mechanical performance of the material. This general trend is confirmed by the behavior of the mechanical loss factor (Fig. 8).
Dielectric behavior
Determination of the electric complex permittivity and its variation against temperature and frequency are among the most important characterization procedures to be performed in dielectric materials [38]. Figure 9 shows the variation of the ε′ for the different PVDF/BaTiO3 samples for several frequencies, obtained at room temperature (Fig. 9a). Figure 9b shows the dielectric constant as a function of filler content as well as the trends obtained with the two applied models.

Variation of ε′ (a) and tan δ (c) as a function of logarithm of frequency of the PVDF and PVDF/BaTiO3 samples and b fitting results for the BSP and Furukawa models for the dielectric permittivity; d represents the conductivity as a function of frequency in a log–log plot
The dielectric constant of the composite samples increases when compared to the polymeric matrix when BaTiO3 particles are introduced [38], especially for the particles with smaller average size. A maximum in ε′ trend is observed for the sample with 10% BaTiO3 (ϕ = 10 nm). For the sample with ϕ = 500 nm the increase in ε′ is not so pronounced and the obtained value for the material with 10% nanoparticle content are smaller than the ones found for the samples with ϕ = 10 nm and 5% ceramic filler, which reveals that the interaction between the ϕ = 10 nm BaTiO3 particles and the polymeric matrix is more effective due to increased interaction area. The specific surface area available in the interaction between the ceramic nanoparticles and the polymeric chains are higher for the smaller particles which increase the space charge distribution and the Maxwell–Wagner–Sillars contribution to the dielectric response due to the dynamics of the charged species in the vicinity of the interfaces [39, 40].
In this way, the increase of the dielectric constant is associated to an increase of the dissipation factor (Fig. 9c) with, in general, increases with increasing filler size and content. The dielectric loss is characterized by an initial decrease with increasing frequency up to 104 Hz (especially relevant for the larger filler size and content sample) due to the hindered mobility of the charged species trapped at the polymer–ceramic interfaces as frequency increases. This behavior can be better understood in the scope to the conductivity contributions. The real part of the conductivity of the dielectric material can be calculated from the dielectric measurements presented in Fig. 9 after Eq. 6:
where ε 0 is the permittivity of free space, ω = 2πf is the angular frequency and \( \varepsilon^{\prime \prime } \left( \omega \right) = \varepsilon^{\prime } \tan \delta \) is the frequency dependent imaginary part of the dielectric permittivity [39].
The \( \sigma^{\prime } \left( \omega \right) \) values for the PVDF polymer matrix and for the PVDF/BaTiO3 composites are presented in Fig. 9 as a function of frequency. The pure α-PVDF polymer shows two well identified regimes, one at lower frequencies up to 104 Hz showing a conductivity mainly dominated by the dc conductivity and a power law dependence for higher frequencies, assigned to the ac contribution to the conductivity [41]. On the other hand the composites have one single regime for the measured conductivity in the 102–107 Hz frequency range, which reveals that the main contribution of the composite conductivity arises from the ac conductivity. It is to notice that for frequencies above 104 Hz the behavior is similar for the pure polymer and the composite with respect to the frequency dependence. The main difference among the samples is therefore the dc contributions, which is suppressed with increasing filler content, due to hindering effects by the fillers to the conductivity of the polymer and a general decrease also of the ac conductivity: the mobility of the ionic species contributing to the ionic conductivity are hindered by the presence of the fillers being also trapped in the filler interfaces (MWS effect) and contributing therefore just to the ac conductivity. The reduction of the conductivity is larger for the smaller filler sizes and the largest filler contents, supporting therefore the MWS contribution to the dielectric response of the composites [39].
The behavior of the dielectric constant was evaluated according to the Bhimasankaram et al. (BSP) [42] and Furukawa et al. [43] theoretical models, explained elsewhere [17]. Briefly, the BSP model is applied to a composite material with 0–3 connectivity in which the system is composed of spherical dielectric material randomly dispersed in a continuous medium. In this model each dielectric sphere is polarized and the dipoles get aligned in the direction of the applied electric field. These dipoles locally modify the electric field in the neighboring region. The effect of local dipolar fields becomes more important for the composites with larger faction of dielectric particles (Eq. 7):
where ε1 is the dielectric constant of the polymer, ε2 is the dielectric constant of the filler, and q the volume fraction of the filler.
Furukawa developed a mathematical expression in order to explain the dielectric behavior of biphasic composites with 0–3 connectivity. This model also assumes that the ceramic particles are spherical and uniformly dispersed in the polymeric matrix. The entire system is dielectrically homogeneous and depends mainly on the dielectric constant of the matrix. The dielectric behavior of the composites is predicted by:
where ε1 is the dielectric constant of the matrix and q the volume fraction of the spherical ceramic particles.
In short, whereas the Furukawa model [43] does not consider the interaction between filler and matrix, this interaction is considered in the BSP model [42].
The obtained results for the fitting with the different mathematical models are presented in Fig. 9b. For the samples with 500 nm average ceramic particle size it was observed that the Furukawa model can fit to the experimental data only for lower concentrations. For the higher concentrations a deviation is observed, being the obtained results higher than the ones predicted by the Furukawa model. On the other hand, the BSP model predicts with very good accuracy the experimental data for the BaTiO3 concentrations and 500 nm particles used in the present study. In this case, the interactions considered in the BSP model are able to resemble the main contributions to the dielectric constant of the composite system. The models presented only consider simple interactions between the filler and the polymeric matrix and consider a homogenous dielectric system, but in the case of the particles with ϕ = 10 nm, as already observed by FTIR and DMA, the interaction between the nanoparticles and the PVDF matrix are more complex, due to the interaction at the interfaces between the BaTiO3 nanoparticles and PVDF molecules, that tend to produce locally oriented CH2–CF2 dipoles that are packed in the TTT configuration characteristic of the β-phase, which increases the dielectric permittivity of the material. This results in a large interface interaction and large contributions of the MWS effects that are not considered by the aforementioned models.
Conclusion
Poly(vinylidene fluoride) (PVDF)/BaTiO3 nanocomposites have been investigated by means of thermal, mechanical, and electrical properties in order to evaluate the effect of filler size and content. Dielectric constant increases significantly when nanometric particles were used as a nucleating agent. This increase is particularly important for the smaller filler sizes and also shows a strong concentration dependence. The dependence of the BaTiO3 particle size in the dielectric properties of the BaTiO3/PVDF composites is ascribed to space charge effects at the interface between BaTiO3 and PVDF, which is especially relevant in for the smaller filler size. The filler matrix interfacial interaction leads, during crystallization, to the nucleation of the β-phase of PVDF. The nucleation process strongly depends on the particle size, being almost independent on the filler content. This trend is also verified for the activation energy of the thermal degradation of the polymer, pointing out again to the importance of the filler size in determining the interaction with the polymer matrix. Smaller nanoparticle act as nucleating agent during polymer phase crystallization whereas the larger particles, with reduced relative interaction area for a given volume concentration, lead to defects formation, as demonstrated by the mechanical properties. Also, increasing filler content leads to more defective structures, as shown by the decrease of the degree of crystallinity of the polymer matrix. As a conclusion, smaller particles lead to larger relative interaction areas between the ceramic filler and the polymer matrix. These interactions are responsible for the main variations observed in the thermal, mechanical, and electrical properties of the composites.
References
Nalwa HS (1991) J Macromol Sci C 31(4):341
Gregorio R, Ueno EM (1999) J Mater Sci 34(18):4489. doi:10.1023/a:1004689205706
Gregorio JR, Cestari M (1994) J Polym Sci B 32(5):859. doi:10.1002/polb.1994.090320509
Lovinger AJ (1982) In: Basset DC (ed) Developments in crystalline polymers. Elsevier, London
Sencadas V, Gregorio Filho R, Lanceros-Mendez S (2006) J Non Cryst Solids 352(21–22):2226. doi:10.1016/j.jnoncrysol.2006.02.052
Galasso FS, Kestigan M (2007) In: Inorganic syntheses. Wiley, New York. doi:10.1002/9780470132456.ch28
Carter CB, Grant Norton M (2007) Ceramic materials: science and engineering. Springer, New York
Jones M (1999) Mechanics of composite materials. Taylor & Francis, Philadelphia
de Mouro MFSFM, de Morais AB, Magalhães MG (2005) Materiais compósitos—materiais, fabrico e comportamento mecânico. Publindústria, Lisboa
Dias CJ, Das-Gupta DK (1996) IEEE Trans Dielectr Electr Insul 3(5):28
Newnham RE, Skinner DP, Cross LE (1978) Mater Res Bull 13(5):525. doi:10.1016/0025-5408(78)90161-7
Muralidhar C, Pillai PKC (1988) Mater Res Bull 23(3):323. doi:10.1016/0025-5408(88)90004-9
Gregorio R, Cestari M, Bernardino FE (1996) J Mater Sci 31(11):2925. doi:10.1007/bf00356003
Hsiang H-I, Lin K-Y, Yen F-S, Hwang C-Y (2001) J Mater Sci 36(15):3809. doi:10.1023/a:1017946405447
Luo XC, Chen L, Chen X, Huang Q (2004) J Mater Sci Technol 20(4):4
Chanmal CV, Jog JP (2008) eXPRESS Polym Lett 2(4):8
Firmino Mendes S, Costa C, Sencadas V, Serrado Nunes J, Costa P, Gregorio R, Gomez Ribelles J, Lanceros-Méndez S (2009) Appl Phys A 96(4):1037. doi:10.1007/s00339-009-5323-y
Costa CM, Firmino Mendes S, Sencadas V, Ferreira A, Gregorio R Jr, Gómez Ribelles JL, Lanceros-Méndez S (2010) J Non Cryst Solids 356(41–42):2127. doi:10.1016/j.jnoncrysol.2010.07.037
Sencadas V, Moreira VM, Lanceros-Méndez S, Pouzada AS, Gregório Filho R (2006) Mater Sci Forum 514–516:5
Sencadas V, Gregorio R, Lanceros-Méndez S (2009) J Macromol Sci B 48(3):514
Salimi A, Yousefi AA (2004) J Polym Sci B 42(18):3487. doi:10.1002/polb.20223
Silva MP, Sencadas V, Botelho G, Machado AV, Rolo AG, Rocha JG, Lanceros-Mendez S (2010) Mater Chem Phys 122(1):87. doi:10.1016/j.matchemphys.2010.02.067
Benz M, Euler WB, Gregory OJ (2000) Langmuir 17(1):239. doi:10.1021/la001206g
Nangia A, Desiraju GR (1999) Chem Commun (7):605
Garetz BA, Matic J, Myerson AS (2002) Phys Rev Lett 89(17):175501
Sencadas V, Costa C, Gómez Ribelles J, Lanceros-Mendez S (2010) J Mater Sci 45(5):1328. doi:10.1007/s10853-009-4086-3
Abraham FF (1970) Science 168(3933):833. doi:10.1126/science.168.3933.833
Falkand M, Knop O (1973) In: Franks F (ed) Water: a comprehensive treatise, vol 2. Plenum, New York, p 55
Sencadas V, Martins P, Pitães A, Benelmekki M, Gómez Ribelles JL, Lanceros-Mendez S (2011) Langmuir 27(11):7241. doi:10.1021/la2008864
Turi E (1997) Thermal characterization of polymeric materials. Academic Press, New York
Sencadas V, Lanceros-Méndez S, Mano JF (2004) Solid State Commun 129(1):5
Flynn JH, Wall LA (1966) J Polym Sci B 4(5):323. doi:10.1002/pol.1966.110040504
Ozawa T (1965) Bull Chem Soc Jpn 38(11):1881
Flynn JH, Wall LA (1967) J Polym Sci B 5(2):191. doi:10.1002/pol.1967.110050211
Mendes SF, Costa CM, Sencadas V, Pereira M, Wu A, Vilarinho PM, Gregorio R Jr, Lanceros-Méndez S (2011) Compos Sci Technol, submitted
Botelho G, Lanceros-Mendez S, Gonçalves AM, Sencadas V, Rocha JG (2008) J Non Cryst Solids 354(1):72. doi:10.1016/j.jnoncrysol.2007.07.012
Avilés M, Ginés J, del Rio J, Pascual J, Pérez-Rodríguez J, Sánchez-Soto P (2002) J Therm Anal Calorim 67(1):177. doi:10.1023/a:1013706501882
Sencadas V, Costa CM, Moreira V, Monteiro J, Mendiratta SK, Mano JF, Lanceros-Mendez S (2005) e-Polymers, art. nº 002
Kremer F, Schonhals A (2003) Broadband dielectric spectroscopy. Springer-Verlag, Berlin, Heidelberg
Van Beek LKH (1960) Physica 26(1):66. doi:10.1016/0031-8914(60)90115-4
Dyre JC (1988) J Appl Phys 64(5):2456
Bhimasankaram T, Suryanarama SV, Prasad G (1998) Curr Sci 74:967
Furukawa T, Ishida K, Fukada E (1979) J Appl Phys 50(7):4904. doi:10.1063/1.325592
Acknowledgements
This work is funded by FEDER funds through the “Programa Operacional Factores de Competitividade—COMPETE” and by national funds by FCT-Fundação para a Ciência e a Tecnologia, project references PTDC/CTM/69316/2006, PTDC/CTM-NAN/112574/2009, and NANO/NMed-SD/0156/2007. The authors also thank support from the COST Action MP1003, 2010 ‘European Scientific Network for Artificial Muscles’. V.S., S. F. M., and C. M. C. thank the FCT for the SFRH/BPD/63148/2009, SFRH/BD/22506/2005, and SFRH/BD/68499/2010 grants, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mendes, S.F., Costa, C.M., Caparros, C. et al. Effect of filler size and concentration on the structure and properties of poly(vinylidene fluoride)/BaTiO3 nanocomposites. J Mater Sci 47, 1378–1388 (2012). https://doi.org/10.1007/s10853-011-5916-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-011-5916-7