
Abstract
Titanium-modified MCM-41 type mesoporous silica materials were prepared by hydrothermal [Ti-MCM-41(HT)], sol–gel [Ti-MCM-41(SG)] and post-synthesis impregnation [TiO2/MCM-41] methods. The materials were characterized and tested as photocatalysts in the oxidative degradation reaction of 2,4,6-trichlorophenol (2,4,6-TCP) in water. The catalysts showed high initial activity. The produced acetate and chloride ions were found to inhibit the degradation reaction. The Ti-MCM-41(HT) sample showed higher overall activity than the Ti-MCM-41(SG) catalyst. One of the probable reasons for this is the difference in the distribution of the active sites that determines the rates of electron (e−)–hole (h+) recombination within the photoactive species. The HT preparation was found to contain silica-bound titania in higher dispersion, while the SG preparation contained also polymerized species with Ti–O–Ti bonds.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chlorophenols (CPs) are persistent and toxic organic compounds, which are resistant to biodegradation and may transform to even more toxic compounds under environmental conditions. CPs represent the most abundant family of industrial toxic materials with their large number of different sources [1]. Due to its bactericidal activities 2,4,6-trichlorophenol (2,4,6-TCP) is used as wood and glue preservative, as antimildew agent for textiles, in pesticide formulations, and in leather tanning and finishing. 2,4,6-TCP has been detected in the emissions from fossil fuel combustion, municipal waste incineration process, and during disinfection of phenol-containing water by chlorination. Trichlorophenols can be found in ground waters, wastewaters and soils, and in ambient air. These toxic and carcinogenic compounds can cause serious environmental contamination affecting public health [2]. The permissible concentration of CPs in drinking water should be below 10 μg/L [3]. For 2,4,6-TCP, a criteria maximum concentration of 1.01 mg/L and a criterion continuous concentration of 0.226 mg/L were determined [4].
Several physical and chemical methods are suitable for the removal of trichlorophenol compounds from wastewaters, such as adsorption [5, 6], coagulation flocculation [7], and reductive dechlorination using zero-valent metals [8, 9]. Advanced oxidation processes (AOPs) based on catalytic or chemical photooxidation are real alternatives of the conventional technologies because almost total degradation of CPs can be achieved without application of high amount of chemicals and generation of waste sludge. AOP techniques, such as UV-ozonization, UV-peroxidation with H2O2 and heterogeneous photocatalytic methods involve reactive free radical species, mainly HO• for non-selective mineralization of organic compounds to harmless end products [3]. Among AOPs, heterogeneous photocatalytic oxidation proved to be an efficient tool for the abatement of organic contaminants. Particularly owing to its excellent photocatalytic activity, TiO2 has been widely studied for the removal of several types of organic pollutants harmful for the environment [10–12]. However, its effective commercial application is still hindered by some disadvantages. Nanoparticles of TiO2 can agglomerate or coalesce very easily forming larger particles resulting in an adverse effect on the catalyst efficiency, while separation and recovery of TiO2 particles from the reactant mixture can be difficult. Immobilization of titania nanoparticles on suitable support can overcome the above problems. Ordered mesoporous silica materials, such as MCM-41 and SBA-15 are promising candidates as supports for titania nanoparticles, since they do not only take the advantage of high surface area and adsorption capacity but also make the catalyst-recovering stage more facilitated [13–20].
Titania can be incorporated into the silica structure by direct or post-synthesis methods resulting in different types of isolated or bulky active sites on the surface of mesopore walls or in the channels, respectively. Sharma et al. [13, 14] have found that 10 wt% TiO2 supported on Al-MCM-41 and SBA-15 showed higher degradation rate for isopruton herbicide than TiO2 itself due to the lack of particle aggregation and light scattering by TiO2. Sadjadi et al. [15] and Anandan [16] investigated TiO2/MCM-41 materials in the photocatalytic degradation of methyl orange and titania immobilized on high surface silica was more efficient than colloidal TiO2 nanoparticles. It was stated that electron-rich MCM-41 surface as a hole scavenger inhibits charge recombination on titania surface. When titanium is isomorphously substituted into the structure of mesoporous silicates, isolated or slightly polymerized titania species as well as anatase nanoclusters can be formed [17–19]. Ti-SBA-15 and Ti-MCM-41 samples were tested in the photocatalytic degradation of dye molecules [17, 18] and it was found that the higher the titanium content the higher the catalytic activity. However, incorporation of titanium into the silica was associated with deterioration of the mesoporous structure; therefore formation of highly dispersed anatase particles on the external surface was found to be responsible for the increasing photoactivity. Wang et al. [19] prepared well dispersed, isolated tetrahedral titanium oxide species by grafting and subsequent hydrolysis of tetraneopentyl titanium complex on the surface of MCM-41. It was evidenced that tetrahedrally coordinated titanium species with oxygen on the surface play a significant role in the gas phase oxidation of ethylene. This was also supported by Orlov et al. [20] who investigated titania-modified SBA-15 materials in degradation of p-chlorophenol and found that variation in the photocatalytic activity is governed by isolated titanium sites.
In this study, Ti-MCM-41 materials, containing different amounts of titanium were prepared via hydrothermal, sol–gel, and impregnation methods. The adsorption and photocatalytic degradation of 2,4,6-TCP were compared on the basis of the properties of the prepared materials. The formed intermediates, acetate and chloride ion was followed to elucidate more about the mechanistic details of the reaction.
Experimental
Catalysts
Ti-MCM-41(HT), having Si to Ti ratio of 25, was synthesized by the hydrothermal method of Trong et al. [21]. Tetraethyl orthosilicate (TEOS) and tetraisopropyl titanate [Ti(OiPr)4] was used as silica and titania sources, respectively. The synthesis mixture had the molar composition of SiO2:0.04 TiO2:0.13 (C16TMA)2O:0.13 (TMA)2O:0.13 (NH4)2O:55 H2O. The obtained solid was washed with distilled water and dried at 333 K. The template was removed by heating it up to 813 K in air at a rate of 1 K/min and keeping this temperature for 5 h.
The Ti-MCM-41(SG) material was prepared with Si to Ti ratio of 10. The room-temperature sol–gel method of Galacho and Carrot [22] was applied. Tetraethylorthotitanate (Ti(OEt)4) was used as titanium source. The molar composition of the synthesis mixture was 1 TEOS:0.1 Ti(OEt)4:0.16 C16TMABr:3 NH3:160 H2O:3.9 EtOH. The suspension of the mixed components was stirred for 2 h and aged overnight at room temperature. The formed precipitate was separated, washed with distilled water until the pH of the washing water remained unchanged, and dried at 313 K. The template was removed from the solid as described above.
The third type of titania-containing MCM-41 sample was obtained by impregnating pure silica MCM-41, prepared by the sol–gel method, with Ti(OiPr)4. 1.5 g of MCM-41 material was suspended in 100 mL 2-propanol and 1.3 g of Ti(OiPr)4 was added to the suspension. The solvent was allowed to evaporate at ambient temperature. The solid residue was dried in air at 383 K for 1 h, heated up to 725 K at a rate of 2 K/min, and kept at this temperature for 3 h. The obtained material, containing 20 wt% TiO2, was denoted to TiO2/MCM-41.
Catalyst characterization
The X-ray powder diffraction (XRD) patterns of the catalysts were recorded by a Philips PW 1810/1870 diffractometer, applying monochromated CuKα radiation (40 kV, 35 mA).
The nitrogen adsorption–desorption isotherms were determined at 77 K using Quantachrom NovaWin2 Automated Gas Adsorption Instrument. The pore size distribution (PSD) of the solid preparations was calculated from the adsorption isotherms according to the BJH approximation [23].
A Morgagni 268D microscope (100 kV; W filament; point-resolution = 0.5 nm) was used to record the transmission electron microscopic (TEM) image of the catalysts.
Diffuse reflectance spectra in the ultra violet-visible region (DR UV–Vis spectra) were used to get information about the electronic state and coordination of titanium in the titanosilicates [24]. The spectra were recorded under ambient conditions using a Jasco V570 spectrophotometer equipped with an integrating sphere. Barium sulfate was used as reference material. FT-IR measurements were carried out by a Nicolet Compact 400 spectrometer, applying the self-supported wafer technique, to obtain spectra from adsorption of pyridine (Py) at 0.07 kPa Py pressure at room temperature. Before Py adsorption, the catalyst wafer was dehydrated in high vacuum at 300 °C. The spectra were normalized to 5 mg/cm2 wafer “thickness”. A BioRad (Digilab) spectrometer was used to record the FT-Raman spectra of the catalysts. It was equipped with Nd:YAG laser, emitting an excitation radiation of 1064 nm wavelength, and with a germanium detector, cooled by liquid nitrogen. Spectra were obtained by the co-addition of 512 individual spectra, each collected at 500 mW laser power and 4 cm−1 resolution. The titanium oxide content of the samples was determined by Shimadzu EDX 720 energy dispersive X-ray spectrometer. Mechanical mixtures of anatase and silica MCM-41 were used to record a calibration curve for the quantitative X-ray analysis.
Photocatalytic measurements
A batch type cylindrical photochemical quartz reactor was used, cooled by circulating water, along the axis of the reactor cylinder. A 300 cm3 of the polluted water was stirred with the suspended catalyst using a magnetic stirrer. The initial concentration of 2,4,6-TCP was 100 ppm. UV lamp at 254 nm wavelength was used as irradiation source. The polluted water was aerated using a bubble distributor and an air input of 100 cm3/min. The catalyst concentration was 0.033 g/L. At certain time intervals 5 cm3 of polluted water was taken from the reactor, filtered and analyzed using HPLC Dionex 202 provided with photodiode array detector at 225 nm wavelength and C18 column (4.6 × 25 cm). The mobile phase was acetonitrile/water in the ratio of 60:40 with a flow rate of 1 mL/min. The concentration of chloride and acetate ions produced during the degradation course was determined by ion chromatography (Dionex-Pac).
Results and discussion
Textural characterization
The XRD patterns and nitrogen adsorption isotherms of the catalysts, shown in Figs. 1 and 2, respectively, are similar to those, usually reported for ordered, hexagonal MCM-41 materials. The compositions and textural characteristics of the catalysts are summarized in Table 1.

XRD patterns of titanium-modified MCM-41 materials

Nitrogen adsorption isotherms of titanium-modified MCM-41 materials
The full width at half maximum (FWHM) of the (100) XRD reflection reflects the long-range order of the titanium-containing MCM-41 catalysts. Transition metal substituted catalysts are generally more disordered than pure silica MCM-41, due to the less flexible metal-oxigen angle than the O–Si–O angle. It can be also observed that the higher the transition metal incorporation, the higher the structure disorder. However, in our case the structure ordering seems to depend rather on the applied preparation method than on the total Ti content of the sample (Table 1). In spite of its lower titanium content Ti-MCM-41(HT) exhibits a broader (100) reflection than the Ti-MCM-41(SG) sample. The relatively narrow (100) reflection of letter sample indicate a well-ordered hexagonal structure (Fig. 1a). The structural regularity of the TiO2/MCM-41 sample is intermediate between the ordered SG and the less-ordered HT preparations. Reflections of anatase were only observed in the XRD pattern of this sample (Fig. 1b). The average particle size of the anatase phase, determined by the Scherer method, was 35 nm.
The N2 adsorption/desorption isotherms are reversible and exhibit a pore-filling step between 0.25 and 0.35 relative pressure (Fig. 2). Among the studied samples, the Ti-MCM-41(SG) sample has the highest pore volume and surface area. The isotherm of this SG prepared sample shows the steepest step of capillary condensation that corresponds to the narrowest PSD. The PSD curves determined by the BJH method (not shown) are in harmony with the properties, revealed by the XRD data, i.e., the pore sizes of Ti-MCM-41(SG) sample are more uniform than that of Ti-MCM-41(HT) sample.
The TEM micrographs show that the morphology of the Ti-MCM-41(HT) and Ti-MCM-41(SG) samples are very much different (Fig. 3). The HT material consists of agglomerated 50–100 nm size particles, having irregular shapes (Fig. 3a). In contrast, the SG preparation comprises of uniform spherical particles, having 200–500 nm diameter. Within the spherical particles a well-ordered parallel channel system can be discerned (Fig. 3b). The sample, prepared by impregnation contains two separate phases: silica MCM-41 with spherical morphology and finely dispersed anatase. The small anatase particles are either bound to the outer surface of the spheres (Fig. 3c), or form separate larger agglomerates (Fig. 3d). The anatase crystallites, observed, were smaller than about 10 nm size, i.e., much smaller than the size deduced from the XRD measurements.

TEM images of titanium-modified MCM-41 materials: a Ti-MCM-41(HT), b Ti-MCM-41(SG), c, d TiO2/MCM-41
Spectroscopic results
The DR UV–Vis spectra of the catalyst preparations and anatase were compared (Fig. 4). The characteristic absorption band of anatase is at 330 nm (Fig. 4d). When the anatase phase is finely dispersed, as in the TiO2/MCM-41 sample, the absorption band appears at shorter wavelength (Fig. 4c). This can be due to quantum size effect of nanoscaled particles or interface interactions, or the combination thereof [11]. The pronounced absorption band of the Ti-MCM-41(HT) catalyst at about 270 nm suggests the presence of higher amount of isolated Ti atoms in penta- or hexa-coordinated environment. These coordination states are common when titanium is surrounded by oxygen atoms and water molecules [24]. The absorption edge of the Ti-MCM-41(SG) sample appears slightly shifted to higher wavelength, substantiating that the absorbing species have more polymerized –Ti–O–Ti– bonds [22]. Hydrolysis of the Ti(OEt)4 during the SG synthesis and partial polymerization of the hydrolysis product can give such species. The relatively high titanium content of the SG synthesis mixture and the ambient temperature of the synthesis facilitate the formation of the above mentioned polymeric species and hinder titanium incorporation into the silica framework.

UV–Vis spectra of titanium-modified MCM-41 compared to anatase
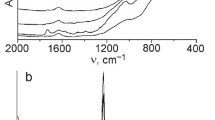
The incorporation of Ti in the silica framework generated Lewis and Brønsted acid surface sites. The species, obtained from adsorption of Py, were studied by infrared spectroscopy to get information about the amount and nature of these sites in the Ti-containing MCM-41 catalysts. The 8a, 8b, 19a, and 19b ring vibrations (νCCN) of Py, which can be physisorbed or H-bonded (HPy), coordinated to Lewis acid sites (LPy) or protonated on Brønsted acid sites (BPy), give absorption bands in the 1700–1400 cm−1 frequency region (Fig. 5a). The bands at about 1607 cm−1 (ν8a), and 1448 cm−1 (ν19b) are assigned to LPy. The protonated Py coordinated to the conjugated base of the solid Brønsted acid (BPy), gives band 1545 cm−1 (ν19b). The weak band at 1446 cm−1 appearing as a shoulder of the 1448 cm−1 band stems from the ν19b vibration of physisorbed Py. The surface silanol groups of the MCM-41 are possible sites of this weak physisorption. The ν19a vibration of any surface bound Py species can give absorption band at 1490 cm−1. The assignment of the 1598 cm−1 band to a specific adsorbed Py species is also dubious. The ν8a, ν8b, and the ν19b vibration of LPy, as well as, the ν19b vibration of HPy can give absorption band at about this frequency. If both kinds of acid sites are present in the sample, due to possible overlapping of the νCCN bands, the ν19a vibrations at about 1450 and 1545 cm−1 are diagnostic for the presence of Lewis and Brønsted acid adsorption sites, respectively. Parallel to the weak 1545 cm−1 the band of the ν8a, ν8b, and the ν19b vibration of BPy can be discerned in the 1650–1630 cm−1 region. Above band assignment is based on [25], and the references therein.

FT-IR spectra of Py, adsorbed on Ti-modified MCM-41 materials (a) and Raman spectra of anatase and the TiO2/MCM-41 material (b)
According to Kataoka and Dumesic [26] Ti–O–Si bridges may generate Brönsted acid sites, if the Ti atoms are in pentahedral/octahedral coordination and hydroxyl groups or water molecules are in their coordination sphere. The dehydrated Ti-containing MCM-41 samples did not show significant Brønsted acidity, only Ti-MCM-41(HT) has weak Brønsted acidic character (Fig. 5a). In the absence of coordinated water or hydroxyl groups the Ti–O–Si species behave as Lewis acid sites. The intensity of the LPy bands is related to the concentration of the Ti–O–Si bonds. In the TiO2/MCM-41 sample such bonds can be formed in the reaction between the hydrolysis product of the Ti precursor, Ti(OiPr)4, and the silanol groups of the MCM-41 surface. The relatively low LPy concentration suggests that only a small fraction of the Ti could participate in this reaction. The results of FT-Raman spectroscopic measurements show that most of the titanium is present in the form of microcrystalline anatase (Fig. 5b).
In contrast, the Ti-MCM-41(HT) sample, having the lowest Ti content among the studied catalysts, shows the highest Lewis site concentration. The extensive incorporation of the Ti into the silica surface is either the consequence or the reason of the found disordered MCM-41 structure of this preparation.
In every aspect, such as the Ti content, the extent of Ti incorporation into the silica framework, and the regularity of the MCM-41 structure, the SG preparation has intermediate characteristics between the two other samples.
Adsorption and photocatalytic decomposition of 2,4,6-TCP
The photocatalytic destruction and the photolysis of 2,4,6-TCP in an aqueous solution was determined as function of the irradiation time (Fig. 6). The photolytic conversion attained 80% in about 120 min. It was much faster in the presence of catalyst, i.e., complete removal of TCP was achieved over the Ti-MCM-41(HT) and the TiO2/MCM-41 catalysts in about 60 min. In the presence of Ti-MCM-41(SG) catalyst the TCP concentration rapidly dropped by about 70% within 10 min. With irradiation time up to 120 min, a slight decrease in the concentration was found (Fig. 6). The variation of the photocatalytic activity can be attributed to several reasons based on the textural and chemical properties of the prepared catalyst.

Photocatalytic degradation of 2,4,6-TCP on titanium-modified MCM-41 catalysts. 300 cm3 aqueous solution, containing 100 ppm 2,4,6-TCP was irradiated by a 6-Watt UV lamp at 254 nm wavelength at a catalyst concentration of 0.033 g/L
It is well known that photocatalytic activity generally depends on the Ti content of the catalyst and also on the coordination environment of titanium. The optimum concentration of titania in post-synthesis modified TiO2/MCM-41 catalysts was found to be 10 wt% [13, 27]. Increasing the titania loading even up to 60 wt% did not increased the photocatalytic degradation efficiency [27]. These finding are in a good agreement with our results. Wang et al. [28] have found that the lower the Ti content, the higher the photocatalytic activity. At low loadings, Ti atoms occupy site-isolated positions in the framework whereas at higher loadings Ti atoms are present in distorted tetrahedral environment or in octahedral coordination. Anpo et al. [29] suggested that charge transfer in excited state of tetrahedrally coordinated titanium oxide species play a significant role in the direct photocatalytic decomposition:
Matsuoka and Anpo [30] generalized this supposition to the photocatalytic process occurring on the transition metal oxides incorporated into the framework of zeolites. Anpo et al. [29] have also found that not only the structure but the dispersion of Ti–O species is decisive. Our UV–Vis and FT-IR results support that Ti-MCM-41(HT) sample has higher amount of isolated titania species than the other two catalysts and because of the lower amount of titania loaded these species can be found in more isolated surroundings.
However, there are many other factors influencing catalytic activity. FT-IR spectrum in Fig. 5 showed that, Ti-MCM-41(HT) catalyst has high amount of Si–OH groups on the surface. Tsai and Cheng [31] reported that the presence of Si–OH groups hinder the electron hole recombination consequently increase the photocatalytic degradation.
Another factor which can control the photoactivity of the prepared catalyst is the surface hydrophilicity. Ti-MCM-41(HT) which is the most hydrophilic catalyst facilitates the adsorption of polar hydrophilic pollutants as reported by Hsien et al. [32] which was confirmed by FT-IR spectra and adsorption data (Fig. 7).

The drop of 2,4,6-TCP concentration as a function of time under the conditions of the photocatalytic reaction (see the legend of Fig. 6), but in the absence of any irradiation
Figure 7 shows the amount of 2,4,6-TCP adsorbed on the catalyst surface as a function of irradiation time. It was found that, Ti-MCM-41(HT) and TiO2/MCM-41 samples had higher adsorption capacity than Ti-MCM-41(SG), however, comparing the initial rates of adsorption, the HT preparation proved to be the highly adsorbent. Several authors [33–35] suggested that there is a clear relationship between substrate adsorption capacity and the photocatalytic activity.
On the other hand, Fig. 8a shows that the production of acetate ions is much faster using Ti-MCM-41(SG) catalyst during the initial irradiation times. The higher acetate concentration decreases the pH value and encourages the formation of a positively charged TiO2 surface which cannot provide hydroxyl groups needed for hydroxyl radical formation and consequently degradation rate is decreased [36]. Figure 8b shows that the production of chloride ion is also much faster using Ti-MCM-41(SG) catalyst during the initial irradiation times. According to Haarstrick et al. [37], the higher chloride ion concentration acts as hydroxyl radical scavenger. Chloride ions can also absorb UV light [38] and therefore decrease the degradation rate.

Formation of acetate ion (a) and chloride ions (b) during the photocatalytic degradation of 2,4,6-TCP in the presence of titanium-modified MCM-41 catalysts
The above facts point toward that despite of the lower absorbability of 2,4,6-TCP on Ti-MCM-41(SG) and the lower amount of excited active sites the reaction takes place much faster on this catalysts but also deactivation proceeds very fast (within 5 min).
Kinetic studies
It was shown above that the chemical environment of the Ti in the MCM-41 material and the photocatalytic activity of the material depends on the preparation method of the material. The relation between the structural and kinetic parameters can give information about the reasons of the found activity differences.
The heterogeneous catalytic photooxidation kinetics is generally described by the Langmuir–Hinshelwood (L–H) kinetic model [39–48]. In this model, the reaction rate (r) is assumed to be proportional with the coverage of the active surface sites by the organic reactant. The proportionality factor is the reaction rate constant (k). It is generally assumed that the adsorption is fast relative to the photocatalytic transformation and, therefore, the surface coverage can be taken as equal with the equilibrium adsorption coverage (θ). The relationship between the θ and the reactant concentration (C) is expressed by the Langmuir equation of the adsorption isotherm. The rate of reaction is given as
where K is the equilibrium adsorption coefficient. When C is very small the Langmuir rate equation approaches the Henry equation and Eq. 2 simplifies to the first order rate equation:
It is often found that the photocatalytic kinetics can be fitted by L–H rate form, however, it was pointed out by Ollis [46, 47] that the very same rate form applies for different mechanisms. Depending on the mechanistic picture k, and K are obtained as apparent coefficients (k app and K app) that may include the rate constants of the assumed elementary sequential adsorption reaction and desorption process as well as the irradiation intensity.
If the reaction proceeds according to first order kinetics the plot of ln(C 0/C) versus t must start from the origin, because at t = 0, C = C 0. If KC ≪ 1 the plot is linear and the apparent rate constant (k app) can be determined from the slope of the line.
The time dependent photocatalytic conversion of 2,4,6-TCP over the Ti-containing MCM-41 catalysts of this study was presented as ln(C 0/C) versus t plots in Fig. 9. After about a 5-min transient period the process corresponds to the first order kinetics. The k app values calculated from the slopes of the ln(C 0/C) versus t plots are given in Table 2. The plot was given also for the Ti-MCM-41(SG) catalyst. However, this plot is not conclusive about the reaction order because this catalyst was virtually inactive after the initial transient period. The C 0−C versus t representation of the data was also linear.

ln(C 0/C) versus irradiation time plots of the Ti-MCM-41 materials
Most probably the transient period is caused by the parallel disappearance of the reactant from the solution due to adsorption and photocatalytic conversion. Interestingly, this transient period is only about 5 min (Fig. 9), while the adsorption equilibrium was found to establish in more than 80 min (Fig. 7). These results are in accordance with the suggestion [46, 47] that the active sites, formed during irradiation, are not the same as the sites of adsorption in the absence of irradiation.
The rapid photocatalytic reaction may cause that no adsorption equilibrium can be established between the solution and the active sites. This can happen due to either non-chemical or to chemical reasons [46, 47]. Because the adsorption process have to balance desorption and reaction processes, both eliminating adsorbed reactant molecules, the coverage can be lower than the equilibrium coverage during the reaction. This is a kinetic reason, which removes the system from being in adsorption equilibrium. Instead of the slow step approximation the pseudo steady state approach was used to treat this kinetic case. Nevertheless, the deduced rate equation has the form of that of Eq. 2 [46, 47].
Reaction intermediates
Figure 10a–c shows the formation and decay of intermediate products as a function of UV irradiation time in the presence of the prepared catalysts. The four intermediates that could be detected by HPLC analysis were hydroxylated products of TCP degradation. This confirms that the primary reactions steps are (i) the hydroxylation of the aromatic ring, (ii) the substitution of chlorine by OH group, and (iii) oxidation of chlorinated hydroquinone to quinone. The compounds were identified as dihydroxytrichlorobenzene (DHTCB), trihydroxydichlorobenzene (THDCB), 3,5-dichlorocatechol (3,5-DCC), and 2,6-dichlorobenzoquinone (2,6-DCBQ). The intermediate products confirm that TCP decomposition on Ti-MCM-41(HT) catalysts proceeds with the mechanism given by Scheme 1 [35, 49, 50]. The same intermediates could be detected during photodegradation of 2,4,6-TCP on TiO2/MCM-41 sample (Fig. 10b).

Formation of dihydroxytrichlorobenzene (DHTCB), 3,5-dichlorocatechol (3,5-DCC), trihydroxydichlorobenzene (THDCB), 2,6-dichlorobenzoquinone (2,6-DCBQ), and benzoquinone (BQ) intermediates during the photocatalytic degradation of 2,4,6-TCP in the presence of Ti-MCM-41(HT) catalyst (a), TiO2/MCM-41 (b) and Ti-MCM-41(SG) catalyst (c)

Proposed schematic photocatalytic degradation mechanism of 2,4,6-TCP on Ti-MCM-41(HT) and TiO2/MCM-41 materials
On the other hand, using Ti-MCM-41(SG) catalyst (Fig. 10c) only 2,6-DCBQ and benzoquinone (BQ) could be observed in very low concentration. This phenomenon indicates that a different pathway of the reaction could be occurred over the catalyst surface. The new pathway can be represented in Scheme 2.

Proposed schematic photocatalytic degradation mechanism of 2,4,6-TCP on Ti-MCM-41(SG)
Figure 11 shows the mineralization of 2,4,6-TCP as a function of irradiation time. Using Ti-MCM-41(HT) and TiO2/MCM-41 the CO2 production increases with time, the complete mineralization can be achieved at 60 min of irradiation. Whereas using Ti-MCM-41(SG) catalyst, the formation of CO2 does not change beyond 35 min of irradiation. i.e., initially the mineralization is faster using Ti-MCM-41(SG) catalyst, but the processes are inhibited parallel with the activity loss.

Formation of carbon-dioxide during the photocatalytic degradation of 2,4,6-TCP in the presence of Ti-MCM-41 catalysts
In summary, analysis of the type and the appearance sequence of the detectable intermediates in the photocatalytic decomposition of TCP over titanium-containing mesoporous silicates are in accordance with the mechanistic picture that proceeds via hydroxyl radicals. It was pointed out, that structural properties, such as Ti coordination surroundings, hydrophobic properties can be with significant influence on the photocatalytic selectivity and efficiency.
Conclusion
Irrespective of the more favorable textural properties of Ti-MCM-41(SG) catalyst compared to that of Ti-MCM-41(HT) and TiO2/MCM-41 sample, it was found that the overall activity of 2,4,6-TCP photodegradation is higher on the latter ones. Photocatalytic degradation using Ti-MCM-41(SG) was much faster at the first 10 min interval not only for the removal of the 2,4,6-TCP but also for its mineralization, however, total deactivation could be observed for the remaining irradiation time. Our results indicate that probably separated tetrahedrally coordinated (in hydrated state penta or octahedral) titanium species can be considered as catalytically active sites in this reaction and the amount, the mode of titanium incorporation, the accessibility of these active sites in the channel system of MCM-41 strongly influences the catalytic activity. Band gap energy of the prepared samples shifted to lower wavelength (compared to pure anatase) as the TiO2 loading decreased. A significant improvement in the photocatalytic activity was obtained with the sample prepared by hydrothermal method and the optimal TiO2 loading was 5 wt%. The increase in TiO2 loading on the support did not enhance the photocatalytic degradation activity. Different methods of preparation, such as sol–gel or hydrothermal can lead to different degradation pathways. It could be also concluded that faster Cl− ion photo removal from CPs can act as an inhibitor by adsorption on the catalyst surface. Furthermore, organic acids accumulation as final photodegradation products can also play the same role.
References
Hazardous Substances Data Base (2001) National Library of Medicine. http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?HSDB
Toxicological profile for chlorophenols (1999) NTIS Accession No. PB91-181545, Agency for Toxic Substances and Disease Registry, Atlanta, GA
Pera-Titus M, Garcya-Molina V, Banos MA, Gimenez J, Esplugas S (2004) Appl Catal B Environ 47:219
Yin D, Hu Sh, Jin H, Yu L (2003) Chemosphere 52:67
Hameed BH, Tan IAW, Ahmad AL (2009) J Hazard Mater 164:1316
Hameed BH (2007) Colloids Surf A Physicochem Eng Aspects 307:45
Ozbelge TA, Ozbeze OH, Baskaya SZ (2002) Chem Eng Process 41:719
Sinha A, Bose P (2009) J Hazard Mater 164:301
Choi J-H, Kim Y-H (2009) J Hazard Mater 166:984
Linsebigler AL, Lu G, Yates JT (1995) Chem Rev 95:735
Hoffmann MR, Martin ST, Choi W, Bahnemann DW (1995) Chem Rev 95:69
Gaya UI, Abdullah AHJ (2008) J Photochem Photobiol C Photochem Rev 9:1
Sharma MVP, Kumari VD, Subrahmanyam M (2008) Chemosphere 72:644
Sharma MVP, Kumari VD, Subrahmanyam M (2008) Chemosphere 73:1562
Sadjadi MS, Farhadyar N, Zare K (2009) Supperlatices Microstruct 46:266
Anandan S (2008) Dyes Pigment 76:535
Jung WY, Baek SH, Yang JS, Lim K-T, Lee MS, Lee G-D, Park SS, Hong S-S (2008) Catal Today 131:437
Mihai GD, Meynen V, Beyers E, Mertens M, Bilba N, Cool P, Vansant EF (2009) J Porous Mater 16:109
Wang X, Lian W, Fu X, Basset J-M, Lefebvre F (2006) J Catal 238:13
Orlov A, Zhai Q-Z, Klinowski J (2006) J Mater Sci 41:2187. doi:10.1007/s10853-006-7184-5
Trong On D, Kapoor MP, Joshi PN, Bonneviot L, Kaliaguine S (1997) Catal Lett 44:171
Galacho C, Ribeiro Carrot MML, Carrot PJM (2007) Micropor Mesopor Mater 100:312
Barrett EP, Joyner LG, Halenda PP (1951) J Am Chem Soc 73:373
Gao X, Wachs IE (1999) Catal Today 51:233
Akçay M (2005) Appl Catal A General 294:156
Kataoka T, Dumesic JA (1988) J Catal 112:66
Artkla S, Kim W, Choi W, Wittayakun J (2009) Appl Catal B Environ 91:157
Wang W, Song M (2006) Micropor Mesopor Mater 96:255
Hu Y, Martra G, Zhang J, Higashimoto Sh, Coluccia S, Anpo M (2006) J Phys Chem B 110:1680
Matsuoka M, Anpo M (2003) J Photochem Photobiol C Photochem Rev 3:225
Sh-J Tsai, Cheng S (1997) Catal Today 33:227
Hsien Y-H, Chang C-F, Chen Y-H, Cheng S (2001) Appl Catal B Environ 31:241
Subramanian V, Pangarkar VG, Beenackers AACM (2000) Clean Prod Process 2:149
Tanaka K, Padermpole K, Hisanaga T (2000) Water Res 34:327
Aal AA, Mahmoud SA, Aboul-Gheit AK (2009) Nanoscale Res Lett 4:627
Barakat MA, Schaeffer H, Hayes G, Ismat-Shah S (2005) Appl Catal B Environ 57:23
Haarstrick A, Kut OM, Heinzle E (1996) Environ Sci Technol 30:817
Bhatkhande DS, Pangarkar VG, Beenackers AACM (2002) J Chem Technol Biotechnol 77:102
Ollis DF (1985) Environ Sci Technol 19:480
Yamazaki S, Tanaka S, Tsukamoto H (1999) J Photochem Photobiol A Chem 121:55
Cunningham J, Sedlák P (1994) J Photochem Photobiol A Chem 77:255
Mills A, Morris S (1993) J Photochem Photobiol A Chem 71:75
Lepore GP, Pant BC, Langford CH (1993) Can J Chem 71:2051
Al-Akabi H, Serpone N, Pelizzetti E, Minero C, Fox MA, Draper RB (1989) Langmuir 5:250
Matthews RW (1988) J Phys Chem 92:6853
Ollis DF (2005) J Phys Chem B 109:2439
Ollis DF (2005) Top Catal 35:217
Murzin D (2006) React Kinet Catal Lett 89:277
Aal AA, Mahmoud SA, Aboul-Gheit AK (2008) Mater Sci Eng C 29:831
Antonaraki S, Androulaki E, Dimotikali D, Hiskia A, Papaconstantinou E (2002) J Photochem Photobiol A Chem 148:191
Acknowledgements
Financial support by the Hungarian Research Fund, OTKA (grant F 61972) and the Egyptian Hungarian Inter-academic Exchange Agreement are greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aboul-Gheit, A.K., Abdel-Hamid, S.M., Mahmoud, S.A. et al. Mesoporous Ti-MCM-41 materials as photodegradation catalysts of 2,4,6-trichlorophenol in water. J Mater Sci 46, 3319–3329 (2011). https://doi.org/10.1007/s10853-010-5219-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-010-5219-4