
Abstract
Near-spherical Y2O2S:Yb3+,Ho3+ nanocrystals (NCs) with an average particle size of 40 nm were synthesized by the coprecipitation method followed by a solid–gas sulfuration technique. The effects of the Ho3+ ion doping concentration on the upconversion luminescence (UCL) property of the NCs was studied through the UCL spectra. Results show that the UCL intensity of Y2O2S:Yb3+,Ho3+ NCs markedly changes with Ho3+ ion concentration, and that the Ho3+ ion concentration quench is observed at 0.25 mol%. This value is only half as much as that in micron Y2O2S prepared by a solid state reaction, which can be attributed to the distinct diffusion mechanism of activator ions in the coprecipitation process. In addition, strong red emissions can be observed in Y2O2S:Yb3+,Ho3+ NCs throughout all Ho3+ doping concentrations used. However, the Ho3+ in micron Y2O2S usually exhibits weak red UCL. Infrared spectra confirm that this result is related to the large vibrational quanta produced by OH− and CO3 2− groups adsorbed onto the surface of NCs. These large vibrational quanta can remarkably increase the probability of 5S2–5F5 and 5I6–5I7 multiphonon relaxation, leading to the enhancement of red emissions arising from 5F5 → 5I8 transitions. The UCL mechanism of the Yb3+–Ho3+ system in nano- and microsized Y2O2S is also discussed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
For decades, nanosized materials have attracted much attention because of their unique and improved optical, electrical, and structural properties compared with conventional bulk materials. Since upconversion luminescence (UCL) was first observed in nanosized Y2O3:Er3+ by Capobianco et al. [1], UCL from near-infrared (NIR) to visible light in nanocrystals (NCs) has elicited increasing interest because of their wide potential applications in various fields, such as infrared detection, molecule recognition, and three-dimensional displays [2–4]. In recent years, the intense demand for biological fluorescence labels with higher luminescent efficiencies and stabilities have resulted in the widespread application of nanocrystalline upconversion phosphors in the field of biomedicine [5–8].
UCL efficiency depends mainly on the host materials. However, biomedical research requires that the host materials not only possess high luminescence efficiency and excellent chemical stability, but also security and innocuity. Y2O2S has been reported as one of the most efficient upconversion hosts because of its advantages, which include a low phonon energy, low symmetry, favorable chemical stability, and nontoxicity; thus, it has attracted considerable attention [9–11]. Currently, studies on nanocrystalline Y2O2S UCL materials focus mainly on the Yb3+, Er3+ co-doped system [9, 10], and studies on Yb3+, Ho3+ co-doped Y2O2S NCs are thus far rare. Our previous study demonstrated that the UCL efficiency of the Y2O2S:Yb3+,Ho3+ system is higher than that of Y2O2S:Yb3+,Er3+ [12], suggesting that Y2O2S:Yb3+,Ho3+ NCs have the potential to be applied as biological fluorescence labels with high sensitivity. Unfortunately, although Hirai et al. [10] and Xing et al. [11] successfully synthesized Y2O2S:Yb3+,Ho3+ NCs by liquid membrane emulsion and homogeneous precipitation methods, respectively, neither of them studied the UCL mechanism of the Yb3+–Ho3+ system in detail.
In this paper, Yb3+, Ho3+ co-doped Y2O2S NCs were prepared by a simple coprecipitation method followed by a solid–gas sulfuration technique, and the special UCL properties and transition mechanism of the Yb3+–Ho3+ system in Y2O2S NCs were studied extensively.
Experimental section
The Y2O2S:Yb3+,Ho3+ NCs were synthesized by the coprecipitation method followed by a solid–gas sulfuration technique. First, Y(NO3)3·5H2O, Ho(NO3)3·5H2O, and Yb(NO3)3·5H2O were weighed to stoichiometric ratios and dissolved in 100 mL of distilled water to form aqueous 0.1 M rare earth-nitrate solutions. The rare earth doping concentrations were 6 mol% for the Yb3+ ion and 0.1, 0.25, 0.5, 0.75, and 1.0 mol% for the Ho3+ ion. A 0.1 M Na2CO3 aqueous solution and PEG4000 were then selected as the precipitating agent and surfactant, respectively. To prepare the Y2O3:Yb3+,Ho3+ precursor, the aqueous nitrate solution was added to 150 mL of the 0.1 M Na2CO3 solution containing 0.15 g PEG4000 under vigorous stirring. Stirring was continued for 30 min. The resultant precipitates were separated centrifugally at 3000 r/min, and washed twice with distilled water and absolute alcohol. Finally, the obtained precipitates were dried at 60 °C for 12 h and calcined at 600 °C for 1 h to produce the Y2O3:Yb3+,Ho3+ white powders.
The sulfuration of oxides was performed via a solid–gas reaction. The white powders produced were placed into a quartz tube and calcined at 700 and 800 °C. During the calcination, sulfur powders were heated at 400 °C to produce sulfur vapor, which was brought into the tube by an N2 gas flow for reaction with the Y2O3 powder. After the reaction was maintained for 1 h, calcination was terminated and only the N2 gas flow was supplied. When the samples had been cooled to room temperature, off-white Y2O2S:Yb3+,Ho3+ NCs doped with different Ho3+ contents were obtained.
A Shimadzu X-ray diffractometer-6000 using Cu Kα radiation was employed to analyze the crystal size and phase of the samples. The tube voltage used was 40 kV, the tube electric current was 30 mA, and the step velocity was 4°/min. The morphology of the samples was determined by transmission electron microscopy (TEM, Tecnai G2 20), and the infrared spectrum was obtained using the potassium bromide technique on a Fourier transform infrared spectrometer (Magna 2IRTM 550, USA) in the wavenumber range of 4000–400 cm−1. A Hitachi F-4500 fluorescence spectrophotometer equipped with a 980 nm laser diode was used to obtain the UCL spectra of the samples; the emission slit was 1.0 nm and the excitation powder was 117 mW. For intensity emission comparisons, all UCL spectra were corrected using a xenon lamp as reference. Dark spectra obtained under the same conditions were also subtracted from all UCL spectra. However, the emission spectra were not corrected for the wavelength dependence of the detection system sensitivity and the refractive index of air (vacuum correction).
Results and discussion
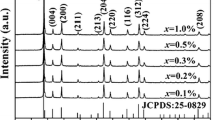
Figure 1 presents the XRD spectra of the Y2O3:6%Yb3+,0.25%Ho3+ precursor material and the samples sulfurized at different temperatures. The Y2O3 precursor obtained crystallizes well and shows a pure cubic phase structure (JCPDS Card No. 74-1828). The considerably broadened diffraction peaks indicate that nanocrystalline Y2O3 particles were obtained. When the precursor is sulfurized at 700 °C for 1 h, the intensities of Y2O3 diffraction peaks decrease gradually, and some weak diffraction peaks for hexagonal Y2O2S structure emerge (JCPDS Card No. 24-1424). After the sulfurized temperature is increased to 800 °C, all diffraction peaks of Y2O3 disappear, and only strong Y2O2S diffraction peaks remain. This result shows that a temperature higher than 800 °C is beneficial for obtaining pure Y2O2S NCs. However, considering that higher temperatures may result in grain growth and particle aggregation, the sulfuration temperature used in this experiment was defined to be 800 °C. According to the Scherrer Formula: \( D = {\frac{K\lambda }{\beta \cos \theta }} \) (where D is the crystal size of the particle, λ is the radiation wavelength of Cu Kα (0.154 nm), and β and θ are the half peak breadth and Bragg diffraction angle of the diffraction peak, respectively), the mean crystal size of the Y2O2S NCs sulfurized at 800 °C is 22.33 nm. Figure 2 shows the TEM image of Y2O2S:6%Yb3+,0.25%Ho3+ NCs sulfurized at 800 °C. The crystals grow relatively well and exhibit a near-spherical shape. The gain size of the sample averages 40 nm and distributes in the range of 25–50 nm. These results are slightly larger than those obtained from XRD measurements, and can be attributed to the slight aggregation of the primary crystals.

XRD spectra of Y2O3:6%Yb3+,0.25%Ho3+ precursor and the samples sulfurized at different temperatures

The TEM image of the Y2O2S:6%Yb3+,0.25%Ho3+ NCs sulfurized at 800 °C
To study the effects of Ho3+ ion content on the UCL properties of Y2O2S:Yb3+,Ho3+ NCs, the UCL spectra of Y2O2S:Yb3+,Ho3+ NCs doped with different concentrations of Ho3+ ions (Yb3+ ion content = 6 mol%) excited at 980 nm LD were obtained, as shown in Fig. 3. The inset in the figure shows the plots of green and red UCL intensities versus the Ho3+ ion concentrations used. Note that the peak positions of Ho3+ characteristic emissions in Y2O2S NCs are consistent with those in bulk Y2O2S [12]. The strong green emissions located at 545 and 548 nm arise from 5F4, 5S2 → 5I8 transitions. The red emissions observed between 650 and 663 nm correspond to the 5F5 → 5I8 transition. The weak NIR emissions located around 759 nm are assigned to 5F4, 5S2 → 5I7 transitions.

The UCL spectra of Y2O2S:Yb3+,Ho3+ NCs doped with different concentrations of Ho3+ ion (Yb3+ content = 6 mol%) excited at 980 nm LD. Inset: The plots of green and red UCL intensities versus the Ho3+ concentrations
Figure 3 shows that the variation of Ho3+ ion concentrations does not affect the peak positions of Ho3+ characteristic emissions. Remarkable influence on the green and red UCL intensities, however, is found. The green UCL intensity improves considerably with increasing Ho3+ concentration, whereas the red UCL intensity exhibits weak enhancement. Both intensities attain maximum levels at an Ho3+ content of 0.25 mol%. When the Ho3+ content is greater than 0.25 mol%, concentration quenching takes place, and the UCL intensities of the green and red emissions begin to gradually decrease. According to the UCL measurement results, the optimal doping concentration of Ho3+ ions for green UCL in Y2O2S NCs is 0.25 mol%. This value is only half as much as that in micron Y2O2S (0.5 mol%) prepared by solid reaction processes [12], and can be ascribed to the distinct diffusion mechanism of activator ions in the coprecipitation process.
During the coprecipitation process, activator ions uniformly distribute into the precursor material produced by the precipitation reaction. While crystallization caused by calcination occurs, activator ions can directly occupy adjacent Y3+ lattices, so that the loss of activator ions is very limited. For the solid reaction, however, activator ions have to diffuse gradually from the surface to the interior of the particles. To accelerate this diffusion, some fusing agent is required to form a mass of grain boundary phases between particles and cause the activator ions to diffuse rapidly into the crystal lattices by a grain boundary diffusion mechanism [13]. According to the EDAX quantitative analysis results, most of the activator ions exist in these grain boundary phases, and the amount of ions which have been doped into the lattices and can operate on upconversion emission does not exceed half of the total doping concentration. Thus, the doping concentration of the activator in materials prepared by solid reactions is usually much higher than the actual requirement.
The red UCL of Ho3+ ions cannot be effectively suppressed in Y2O2S NCs throughout all the Ho3+ doping concentrations used in this experiment, especially since Ho3+ ions in micron Y2O2S typically exhibit considerably weak red UCL [12]. The relatively strong red UCL observed in Y2O2S NCs can be ascribed to the high surface activity of the NCs. The amount of NC surface atoms can highly increase with decreases in crystal size. The deficient coordination and high surface energy cause these surface atoms to possess high activity and show weak stability. To decrease surface energy, a sizeable amount of H2O and CO2 are absorbed onto the NC particle surfaces to form chemisorbed OH− and CO3 2−. These findings are demonstrated by the IR spectrum of Y2O2S:6%Yb3+,0.25%Ho3+ NCs sulfurized at 800 °C (Fig. 4). The broad absorption bands around 3450 and 1660 cm−1 can be assigned to the stretching and bending vibrations of O–H; the bands around 1530 and 1390 cm−1 result from C–O asymmetrical stretching vibrations; the peak that appears at 1140 cm−1 can be assigned to C–O symmetric stretching vibrations; the peaks at 993 and 725 cm−1 correspond to C–O and O–H deformation vibrations; and the strong peak at ~ 500 cm−1 can be assigned to the vibrations of Y–O and Y–S. These large vibrational quanta (3450–1140 cm−1) can considerably influence the transition mechanism of the Yb3+–Ho3+ system in Y2O2S NCs. To further understand this mechanism, the UCL intensities of Y2O2S:6%Yb3+,0.25%Ho3+ NCs excited by 980 LD with different powers were measured.

IR spectrum of Y2O2S: 6%Yb3+,0.25%Ho3+ NCs sulfurized at 800 °C for 1 h
The UCL intensity (I UCL ) is proportional to some power n of the incident excitation power (I P ) so that
where the superscript n = 1, 2, 3… represents the number of pump photons required to populate the emitting states. The integrated intensities calculated from the area under the green, red, and NIR emissions in Y2O2S:6%Yb3+,0.25%Ho3+ NCs as a function of the pump intensity are shown in Fig. 5. The n values are calculated to be 2.03, 2.07, and 1.82 for the green, red, and NIR emission bands, respectively. These indicate that two IR photon absorptions are involved in the emission processes. According to the abovementioned results, the transition mechanism of the Yb3+–Ho3+ system in Y2O2S NCs can be built (Fig. 6).

The integrated intensities of Ho3+ ions green, red, NIR emission in Y2O2S:6%Yb3+,0.25%Ho3+ NCs as a function of the pump intensity

The transition mechanism of Yb3+–Ho3+ system in Y2O2S NCs
Under 980 nm LD excitation, the Ho3+ ions in the ground 5I8 state are excited to 5F4, 5S2 states via two successive energy transfers from the Yb3+ ions in the 2F5/2 state. Transitions from the 5F4, 5S2 states to the 5I8 and 5I7 states then yield green and NIR emissions, respectively. These transition processes are the same as those in micron Y2O2S. However, the red UCL observed from 650–663 nm shows a large difference in the transition mechanisms of the system under study and micron Y2O2S. The energy gaps (ΔE) of 5S2-5F5 and 5I6-5I7 are 2660 and 3220 cm−1, respectively. According to the energy gap law, efficient multiphonon relaxation can take place only when ΔE is equal to or less than 4–5 times the highest energy phonons in the system. For micron Y2O2S co-doped with Ho3+ and Yb3+ ions, its only intrinsic phonon energy (about 520 cm−1) can hardly bridge the large energy difference between the levels mentioned above, and no other phonons are available for its use. Thus, the 5F5 red emission state of Ho3+ in micron Y2O2S is populated mainly by infrequent 5F4/5S2 → 5I7 radiative transitions and the subsequent 5I7 → 5F5 excited-state absorption process (Route 1). Therefore, only weak red UCL can be observed in micron Y2O2S.
For the Y2O2S NCs, however, as mentioned previously, the OH− and CO3 2− groups chemisorbed onto the surface of the NCs can produce large vibrational quanta of 1500 and 3350 cm−1. These quanta can easily bridge the energy gaps of 5F4/5S2-5F5 and 5I6-5I7, and accordingly increase the probability of 5F4/5S2 → 5F5 and 5I6 → 5I7 multiphonon relaxation. Thus, except for Route 1, the population route of the 5F5 state in the NCs should also include the process of 5F4/5S2 → 5F5 multiphonon relaxation (Route 2) and the process of 5I6 → 5I7 multiphonon relaxation, followed by 5I7 → 5F5 excited-state absorption (Route 3). Routes 2 and 3 can increase the population of 5F5 red emission states to a large extent; hence, stronger red emissions can be observed in nanosized Y2O2S. Similar phenomena can also be observed in other nanomaterials doped with rare earth metals [3, 14]. Vetrone et al.’s report revealed that vibrations from OH− and CO3 2− groups can be still observed even after heating NCs to 1000 °C for 65 h [3].
The prepared Y2O2S:Yb3+,Ho3+ NCs exhibit bright green emission in the daytime that is visible to the naked eye at excitation powers as low as 25 mW (IG/IR = 2.53). Because previous studies on green UCL in NCs focused mainly on the Yb3+, Er3+ co-doped system, for purposes of comparison, Y2O2S:Yb3+, Er3+ NCs were also prepared using the procedure described in reference literature [15]. To obtain approximate IG/IR values, the doping concentrations of both Yb3+ and Er3+ions selected for this paper were 0.5 mol% (IG/IR = 2.09; the higher the Yb3+ or Er3+ ion content, the lower the IG/IR value is [15]). Figure 7 shows that in equivalent excitation conditions, the relative UCL intensities of Y2O2S:Yb3+,Ho3+ and Y2O2S:Yb3+, Er3+ NCs can be directly evaluated according to their UCL spectra. The green UCL intensities of the Yb3+–Ho3+ system are all three times higher than those of the Yb3+-Er3+ system throughout all the powers measured (inset, Fig. 7).

The comparison of UCL intensities for Y2O2S:Yb3+,Ho3+ and Y2O2S:Yb3+, Er3+ NCs under 980 nm infrared excitation. Inset: The plots of green UCL intensities of Y2O2S:Yb3+,Ho3+ and Y2O2S:Yb3+, Er3+ NCs versus the excitation powers
Conclusion
Near-spherical Y2O2S:Yb3+,Ho3+ NCs with hexagonal structure were synthesized using the coprecipitation method followed by a solid–gas sulfuration technique. The crystal size of the NCs average 40 nm and are distributed from 25 to 50 nm. Because the coprecipitation method can ensure the uniform distribution of activator ions in the precursor material, the activator ions can directly occupy the adjacent Y3+ lattices during the crystallization process, thereby effectively decreasing the loss of Ho3+ ions. Consequently, the optimal doping concentrations of Ho3+ ions in Y2O2S NCs is 0.25 mol%—only half as much as that in micron Y2O2S. The UCL spectra indicate that the Ho3+ content can considerably influence the UCL efficiency of Y2O2S NCs. The large vibrational quanta produced by OH− and CO3 2− groups adsorbed onto the surface of the NCs can remarkably increase the probability of 5S2–5F5 and 5I6–5I7 multiphonon relaxation. These relaxation processes greatly enhance the population of 5F5 red emission levels, thereby producing stronger red emissions from nanosized Y2O2S than from micro Y2O2S. These results are of importance in research studies that aim to determine nanosized upconversion biological fluorescence labels with high sensitivity.
References
Capobianco JA, Vetrone F, D’Alesio T, Tessari G, Speghini A (2000) Phys Chem Chem Phys 2(14):3203
Wang XF, Xiao SG, Yang XL, Ding JW (2008) J Mater Sci 43(4):1354. doi:10.1007/s10853-007-2266-6
Konishi T, Shimizu M, Kameyama Y, Soga K (2007) J Mater Sci Mater Electron 18(S1):183
Zako T, Nagata H, Terada N, Sakono M, Soga K, Maeda M (2008) J Mater Sci 43(15):5325. doi:10.1007/s10853-008-2776-x
Hampl J, Hall M, Mufti N, Yao YM, MacQueen DB, Wright WH, Cooper DE (2001) Anal Biochem 288(2):176
Rijke F, Zijlmans H, Li S, Vail T, Raap AK, Niedbala RS, Tanke HJ (2001) Nat Biotechnol 19(3):273
Zijlmans HJMAA, Bonnet J, Burton J, Kardos K, Vail T, Niedbala RS, Tanke HJ (1999) Anal Biochem 267(1):30
Tan MQ, Ye ZQ, Wang GL, Yuan JL (2004) Chem Mater 16(12):2494
Pires AM, Serra OA, Davolos MR (2004) J Alloys Compd 374(1–2):181
Hirai T, Orikoshi T (2004) J Colloid Interface Sci 273(2):470
Xing MM, Cao WH, Pang T, Ling XQ (2009) Solid State Commun 149(23–24):911
Luo XX, Cao WH (2007) Mater Lett 61(17):3696
Luo XX, Cao WH, Xiao ZG (2006) J Alloys Compd 416(1–2):250
Rosa EDL, Salas P, Desirena H, Angeles C, Rodríguez RA (2005) Appl Phys Lett 87(24):241912
Zhong HY, Cao WH (2009) J Funct Mater 40(6):896
Acknowledgements
The authors would like to acknowledge the financial support from the National Natural Science Foundation of China (No. 60979003), the Foundation of University Research Program by Liaoning Educational Committee (No. 2009A095), New Century Educational Talents Plan of Chinese Education Ministry (No. NCET-10-0171), and Basic Research Training Foundation of China (No. 2009JC22).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fu, Y., Cao, W., Peng, Y. et al. The upconversion luminescence properties of the Yb3+–Ho3+ system in nanocrystalline Y2O2S. J Mater Sci 45, 6556–6561 (2010). https://doi.org/10.1007/s10853-010-4744-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-010-4744-5