
Abstract
Butyl methacrylate/hydroxyethyl methacrylate (HEMA) copolymeric fiber was prepared by gelation-spinning in twin screw extruding machine, the swelling behavior, absorptive kinetics, and crystallization behavior were investigated, finally the morphology was observed by SEM. The results show that absorptive rate can be quickened and absorbency can be increased with an increase in mass fraction of HEMA. Under the same condition, the fiber has greater capability to absorb chloroform and trichloroethylene, but it has relatively weaker capability to absorb toluene. Additionally, the fiber can selectively absorb toluene from mixed system with high efficiency during a short time. When mass fraction of HEMA is 10 wt% or 15 wt%, Eq. 7 can well describe absorptive kinetics. The polymer melt cannot assume a crystalline structure under the cooling shaping condition during the spinning process, but the melt can form crystals in the cooling process after the fiber has absorbed chloroform for 24 h. The surface becomes coarser and coarser, and the cross section becomes irregular with an increase in mass fraction of HEMA, what is more, number of cavities on the surface and cross section increases as mass fraction of HEMA increases.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, pollution induced by accidental and deliberate releases of organic matter (such as chloroform, toluene, trichloroethylene, etc.) during transportation and storage has been noted as a major cause for environmental pollution. The adverse impacts to ecosystems and the long-term effects of environmental pollution by these and other releases call for an urgent need to develop a wide range of materials for cleaning up organic matter from its impacted areas. A wide range of materials for release remediation have actually been employed, such as dispersants, absorbents, solidifiers, booms, and skimmers [1]. As a type of absorbent material, absorptive resin is attractive for some applications because of the possibility of collection and complete removal of the organic matter from its spill site [2–6]. However, the morphology has set up obstacles for further application due to the fact that almost all the absorptive resins are synthesized as granular polymer by various polymerization methods. Under the circumstances, functional fiber absorbing organic matter should emerge as the times require on account of some good properties including large surface area, high rate of uptake, high uptake capacity, retention over time, organic matter recovery from absorbents, reusability, and textile processability, but traditional resin is synthesized by the copolymerization of hydrophobic monomers with chemical crosslinker, so three-dimensional network is constructed among macromolecules to make the resin not dissolve into organic solvent and not be melted. In this way, it is impossible for absorptive resin to prepare function fiber by traditional methods, such as wet-spinning and melt-spinning, so there are almost no other reports except those from our research team on the functional fiber based on absorptive resin. In order to treat spilled organic matter and develop the advantages of functional fiber, our research team successfully prepared absorptive functional fiber for the first time by using the technology of semi-interpenetrating polymer network and the method of wet-spinning [7]. Unsatisfactorily, the functional fiber has the disadvantage of low strength, in addition, wet-spinning can again cause environmental pollution because of plentiful residual solvent in coagulation bath. For the sake of improving strength of fiber and reducing environmental pollution, a novel absorptive resin which is endowed with the capability to absorb organic matter and the property of melt processing under the swelling state was synthesized, and then the resin was made into fiber by the method of gelation-spinning in plunger spinner or twin screw extruding machine, respectively [8–11].
The study was designed to investigate the absorptive property and kinetics, crystallization behavior, and relationship between morphology and property.
Experimental
Materials
Butyl methacrylate (BMA) was purchased from Sinopharm Chemical Reagent Co., Ltd.; hydroxyethyl methacrylate (HEMA) was from Tianjin Chemical Research Institute and used directly; benzoyl peroxide (BPO) was purchased from China National Pharmaceutical Group Corporation Shanghai Chemical Reagent Company and used after recrystallization according to the literature [12]; poly(vinyl alcohol) (PVA) was purchased from Tianjin Chemical Reagent Co., Ltd.; N,N-dimethyl formamide (DMF) was from Tianjin Bodi Chemical Co., Ltd.; Chloroform was from Tianjin Chemical Reagent Co., Ltd.; Sodium hydroxide was from Tianjin Fengchuan Chemical Reagent Science and Technology Co., Ltd.; Sodium chloride was from Tianjin Tanggu Chemical Reagent Factory.
BMA refinement
First, 500 mL BMA and 500 mL deionized water were stirred in a 2000 mL three mouth flask at room temperature for 5 min. Afterward, the mixture was kept for several minutes until two layers were formed, then the BMA was obtained. Second, the obtained BMA and 500 mL 10 wt% sodium hydroxide aqueous solution were stirred in another three mouth flask at room temperature for 5 min, the BMA was again obtained by separating from sodium hydroxide aqueous solution, and then the BMA was washed with 500 mL 10 wt% sodium chloride aqueous solution for 5 min. After separating from sodium chloride aqueous solution, the BMA was washed by deionized water for several times until the BMA became neutral. Finally, the BMA was purified by distillation under reduced pressure after separating from deionized water.
Preparation of BMA/HEMA copolymer
PVA (MPVA/Mmonomer = 0.5%), as suspension stabilizer, was dissolved in 100 mL deionized water in a 250 mL beaker, and then the PVA solution and 4700 mL deionized water were put into a polymerization kettle. Afterward, a mixture of BMA and HEMA, which total volume was equal to 1600 mL while mass fraction of HEMA in monomer feed ratio was assigned to 0, 5, 10, or 15 wt%, was added in a 3,000 mL beaker, and then the mixture of BMA, HEMA, and BPO was stirred until the BPO was dissolved after BPO (MBPO/Mmonomer = 0.5%), as initiator, was added in the 3,000 mL beaker. Finally, the solution of BMA, HEMA, and BPO was put into the polymerization kettle and stirred to react at 85 °C for 4 h under a nitrogen atmosphere, and then the temperature was increased to 95 °C to make the system continue to react for 2 h, white granular copolymer was obtained after the product was washed and dried in vacuum oven at 60 °C for 72 h.
Preparation of functional fiber
A weighed quantity of dried copolymer was mixed with quantitative DMF, and then the mixture was deposited in a closed container for 48–72 h to enable the copolymer to be swollen adequately at room temperature. Finally, the functional fiber was prepared by the method of gelation-spinning in a twin screw extruding machine. During the spinning process, first, the melt was coagulated in water bath to form as-spun fiber, and then the as-spun fiber was stretched in water bath at 85–95 °C, second, the fiber was processed by following order: winding, cutting off, washing by distilled water and drying at room temperature, eventually, the fiber was preserved for test and analysis.
The spinning parameters are as follows: the heating temperature for the first zone, the second zone, the third zone, the fourth zone, the fifth zone, the sixth zone, and the seventh zone were 90–125 °C, 105–135 °C, 105–135 °C, 105–140 °C, 110–145 °C, 110–145 °C, and 115–150 °C, respectively. In addition, melt temperature and spinneret temperature were 125–145 °C and 80–150 °C, respectively. The frequency for main engine, feeding machine, and melt pump were 6.3–7.0, 4.3–5.1, and 4.0–9.0 Hz, respectively.
Preparation of toluene/water mixed system
A determined volume of toluene (the density is 0.867 g/mL) was added into a 100-mL volumetric flask with a microsyringe, then deionized water was added into the volumetric flask up to its calibration. Finally, the volumetric flask was oscillated at the rate of 100r/min at room temperature in a water bath constant temperature oscillator to make toluene disperse.
Characterization
Organic matter absorbency
A weighed quantity of fiber was immersed into the selected organic matter at room temperature until equilibrium was reached. The equilibrium was determined by measuring the organic matter absorbency every 1 h until it reached a limiting value (the difference of the adjacent values was between negative 5 mg and positive 5 mg). The residual organic matter was removed by dropping for 10 min during the every process of measure. The organic matter absorbency, Q, was determined by weighing the swollen gel and calculated according to the following equation:
where G1 is weight of swollen gel and G0 is weight of dried fiber.
The remaining ratio
After the swelling test, each sample was dried in a vacuum oven at 50 °C for about 3 days, and then the sample was weighed. The remaining ratio, R, was calculated according to the following equation:
where Gb is weight of dried sample after swelling test and Ga is weight of dried fiber before swelling test.
UV analysis
A SHIMADZU UV-2401PC ultraviolet spectrophotometer was used to determine the concentration of residual toluene in the mixed system. The wavelength used in the UV spectrophotometer was 261 nm.
Selective absorption
A determined amount of fiber was put into the volumetric flask containing toluene/water mixed system, and then the volumetric flask was oscillated at the rate of 100r/min at room temperature in a water bath constant temperature oscillator until test time was reached. The absorbency and the absorptive efficiency at time t were calculated by using the following equations:
where Qt and Et are the absorbency and the absorptive efficiency at time t; C0 is the initial concentration and is equal to 0.867 g/L; Ct is the concentration of residual toluene at time t; V is the volume of mixed system and is equal to 100 mL; W is the mass of fiber used in each experiment and is equal to 0.1 g.
The concentration of residual toluene at time t was calculated according to the following equation:
where Ct is the concentration of residual toluene at time t; Cs is the concentration of standard liquid and is equal to 0.867 g/L; At is the absorbency of mixed system at time t; and As is the absorbency of standard liquid.
WAXD analysis
The crystallization ability of fiber was investigated by using an instrument BRUKER AXS D8 DISCOVER with GADDS wide angle X-ray diffractometer (WAXD). Diffractometer with copper cathode was operated in reflectance mode at Cu Kα1 wavelength (λ = 1.5406 Å), 40 kV and 40 mA. Measurement was performed with 2θ scale from 10° to 44.5° at a scanning rate of 4°/min.
DSC analysis
A NETZSCH DSC 200F3 differential scanning calorimeter equipped with a cooling device was used to characterize the thermal behavior of the samples. All measurements were carried out under nitrogen atmosphere at a heating/cooling rate of 10 °C/min. For that purpose, a first heating stage was performed from 10 °C to 200 °C and maintained at 200 °C for 3 min to make the sample be melted adequately, followed by a subsequent cooling at the same controlled rate. In order to remove any previously existing thermal history on the samples, the thermal behavior was characterized only by the second heating stage (also from 10 to 200 °C).
Morphology observation
Surface and cross section morphology were observed by a FEI QUANTA200 scanning electronic microscope (SEM). Surface and cross section obtained by brittle fracture in liquid nitrogen were coated with gold by an electro-deposition method to impart electrical conduction before recording the SEM micrographs.
Results and discussion
Swelling behavior
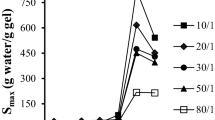
Chloroform, toluene, and trichloroethylene, as volatile and harmful matter, which can cause serious environmental pollution, whether the fiber has great capability to absorb them, so the absorption for these organic matters was investigated in this work and the results are shown in Fig. 1. As Fig. 1a–c shows, for the fiber prepared by BMA homopolymer, the absorbency for chloroform, toluene, or trichloroethylene decreases with the increase of time due to linear structure, and it is inclined to constant value of −1 g/g after 3 or 2 h, which indicates most of BMA homopolymer fiber can dissolve into chloroform, toluene, or trichloroethylene. For BMA/HEMA copolymer fiber, the absorbency increases as time increases, as shown in Fig. 1a–c. The phenomenon demonstrates that network structure can be constructed among macromolecules after BMA finishes copolymerization reaction with HEMA, in accordance with the literatures [9, 13]. In addition, the fiber has different capability to absorb these organic matters as mass fraction of HEMA in monomer feed ratio changes, for example, the absorbency for all of the tested organic matters increases systematically with an increase in mass fraction of HEMA for the same absorptive time, which indicates that the fiber can obtain more perfect and suitable network structure with increasing mass fraction of HEMA. However, the fiber shows different absorbency as organic matter changes, at the same absorptive time, when mass fraction of HEMA in monomer feed ratio is equal to 15 wt%, the fiber shows higher absorbency for chloroform or trichloroethylene, but shows lower absorbency for toluene, which indicates that the fiber has great capability to absorb chloroform and trichloroethylene. It is well known that absorptive functional fiber, as a type of polymer with low crosslinking degree, is composed of soluble and insoluble parts. In general, the remaining ratio is related to proportion of insoluble part and can be used to describe crosslinking density of network structure [14]. For this work, Fig. 2 shows that the remaining ratio increases with the increase of mass fraction of HEMA. The phenomenon again demonstrates that crosslinking density increases as mass fraction of HEMA increases, which is consistent with the fact shown by Fig. 1a–c. Furthermore, for the same mass fraction of HEMA, the remaining ratio in chloroform or trichloroethylene is higher than the one in toluene, which also causes that the fiber has great capability to absorb chloroform and trichloroethylene. Finally, it is well known that the fiber should be used to clean up organic matter spilt in water resource for practical application, so the capability to absorb toluene from mixed system containing toluene and water was investigated, and the results are shown in Fig. 3a–c. It can be seen that the concentration of toluene in mixed system first decreases then increases with prolonging time, and absorbency and absorptive efficiency first increase then decrease with increasing time, which indicates that the fiber can absorb toluene from mixed system with high efficiency during a short time.

Variation of absorbency with absorptive time; mass fraction of HEMA in monomer feed ratio is labeled as follows: (a) 0 wt%, (b) 5 wt%, (c) 10 wt%, (d) 15 wt%; a for chloroform as the tested organic matter, b for toluene as the tested organic matter, and c for trichloroethylene as the tested organic matter

Variation of the remaining ratio with mass fraction of HEMA in monomer feed ratio; chloroform, toluene, and trichloroethylene as the tested organic matters

Effect of absorptive time on selective absorption behavior; mass fraction of HEMA in monomer feed ratio is equal to 15 wt%; toluene as the tested organic matter
Absorptive kinetics
It is well known that absorption rate should be equal to desorption rate when the absorption reaches equilibrium state, so the following equation can be obtained:
Rearranging Eq. 6 results in:
where Qt is the absorbency at time t, Qmax is the maximum absorbency, a, as the absorptive coefficient, is equal to k1/k−1, k1, and k−1 are the absorption rate constant and desorption rate constant, respectively.
In addition, according to Wu and Zhou [15], the absorptive kinetics of the crosslinked polymer can be described by the following experimental equation, namely, second-order kinetics equation:
where K is the equilibrium rate constant of second-order absorption.
If a in Eq. 7 is denoted by KQmax, Eq. 7 is similar to Eq. 8, so Eq. 7 also can be used to describe absorptive kinetics. Plotting t/Qt versus t yields a straight line, as shown in Fig. 4. The values of R2 listed in Table 1 suggest that Eq. 7 is more suitable for describing absorptive kinetics of the fiber prepared as mass fraction of HEMA is 10 or 15 wt%. The parameters in Table 2 indicate that the fiber prepared as mass fraction of HEMA is 10 or 15 wt% can absorb a large amount of chloroform at a quicker rate. However, Table 3 shows that Eq. 7 is the most suitable for describing absorptive kinetics of the fiber prepared as mass fraction of HEMA is 15 wt%, but the Eq. 7 cannot be used to describe absorptive kinetics of the fiber prepared as mass fraction of HEMA is 5 wt%. The phenomenon should be attributed to a lot of soluble part caused by weaker network structure, in accordance with the fact based on the remaining ratio.

Relation curves of t/Qt versus t obtained by linear fit; chloroform as the tested organic matter; mass fraction of HEMA in monomer feed ratio is labeled as follows: (a) 5 wt%, (b) 10 wt%, (c) 15 wt%; solid line for fit curve and dot line for experimental curve
Crystallization behavior
The results analyzed by WAXD and DSC are shown in Figs. 5 and 6. Figure 5 shows that there are no obvious diffraction peaks except the peaks caused by amorphous region within the range of 10–25°, which indicates that the melt cannot assume a crystalline structure under the cooling shaping condition. In fact, the fiber must be amorphous because large numbers of crosslinking points can inhibit chain segments and macromolecular chain from movement under the cooling shaping condition to cause the macromolecules not to arrange in good order, which is consistent with the fact indicated by curve a in Fig. 6a, b. However, the melt can form crystals after the fiber has absorbed chloroform for 24 h, as shown by curve b in Fig. 6a, b. This phenomenon demonstrates that a lot of crosslinking points are destroyed during the absorptive process, and macromolecules can easily rearrange in good order to form crystals during the cooling process.

Equatorial WAXD diffraction patterns of fiber; mass fraction of HEMA in monomer feed ratio is labeled as follows: (a) 0 wt%, (b) 5 wt%, (c) 10 wt%, (d) 15 wt%

DSC thermograms of fiber; (a) for cooling thermograms, (b) for heating thermograms; mass fraction of HEMA in monomer feed ratio is 15 wt%; (a) dried sample before absorption, (b) dried sample after absorbing chloroform for 24 h
Morphology
The surface and cross section morphology of fiber are shown in Fig. 7. It can be found that the surface for BMA homopolymer fiber displays a regular shape. However, the surface becomes coarser and coarser with an increase in mass fraction of HEMA, especially when mass fraction of HEMA is 15 wt%, the surface looks like withered bark. In addition, the cross section becomes irregular with increasing mass fraction of HEMA, what is more, number of cavities on the cross section increases as mass fraction of HEMA increases. These phenomena demonstrate that spinnability of corresponding polymer is weakened with increasing mass fraction of HEMA. However, organic matter can easily permeate into the fiber through the cavities on surface, and organic matter can conveniently diffuse by internal cavities, so absorptive rate can be quickened and absorbency can be increased with an increase in mass fraction of HEMA, which is well in accordance with the fact based on Fig. 1.

SEM photographs of surface and cross section; the percentage denotes mass fraction of HEMA in monomer feed ratio
Conclusions
The network structure becomes more perfect and the corresponding fiber is endowed with greater capability to absorb organic matter with an increase in mass fraction of HEMA. Under the same condition, the fiber has greater capability to absorb chloroform and trichloroethylene, but it has relatively weaker capability to absorb toluene. Additionally, the fiber can selectively absorb toluene from mixed system with high efficiency during a short time. Equation 7 is more suitable for describing absorptive kinetics of the fiber prepared as mass fraction of HEMA is 10 wt% or 15 wt%, but cannot be used to describe absorptive kinetics of the fiber prepared as mass fraction of HEMA is 5 wt%. The polymer melt cannot assume a crystalline structure under the cooling shaping condition during the spinning process, but the melt can form crystals after the fiber has absorbed chloroform for 24 h. Spinnability of corresponding polymer is weakened, but absorptive rate can be quickened and absorbency can be increased with an increase in mass fraction of HEMA.
References
Adebajo MO, Frost RL, Kloprogge JT, Carmody O, Kokot S (2003) J Porous Mat 10:159
Wu B, Zhou MH (2009) Waste Manage 29:355
Atta AM, Arndt KF (2005) J Appl Polym Sci 97:80
Shan GR, Xu PY, Weng ZX, Huang ZM (2003) J Appl Polym Sci 89:3309
Atta AM, EI-Ghazawy RAM, Farag RK, EI-Kafrawy AF, Abdel-Azim AAA (2005) Polym Int 54:1088
Atta AM, EI-Ghazawy RAM, Farag RK, EI-Kafrawy AF, Abdel-Azim AAA (2006) React Funct Polym 66:931
Feng Y, Xiao CF (2006) J Appl Polym Sci 101:1248
Xu NK, Xiao CF, Zhang Y, Feng Y (2008) Polym Mater Sci Eng 24:143
Xu NK, Xiao CF, Song Z (2008) Chem J Chinese U 29:1677
Xu NK, Xiao CF, Feng Y, Song Z, Zhang ZY (2009) Polym Plast Technol 48:716
Xu NK, Xiao CF, Song Z (2009) Acta Polym Sin 4:317
He WD (2003) In: Kong QY (ed) The chemical experiment of polymer. University of Science and Technology of China Press, HeFei
Zhu CQ (1985) In: Geng LC (ed) Acrylate and their polymer-II. Chemical Industry Press, Beijing
Jang J, Kim BS (2000) J Appl Polym Sci 77:914
Wu B, Zhou MH (2009) J Environ Manage 90:217
Acknowledgements
The authors acknowledge the financial support provided by the National Nature Science Foundation of China (Project number: 50673077).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xu, N., Xiao, C. Swelling and crystallization behaviors of absorptive functional fiber based on butyl methacrylate/hydroxyethyl methacrylate copolymer. J Mater Sci 45, 98–105 (2010). https://doi.org/10.1007/s10853-009-3897-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-009-3897-6