
Abstract
Fe–4.6 wt% B alloy was synthesized via electro-deoxidation of the mixed oxide precursor. The oxides, Fe2O3 and B2O3, mixed in suitable proportions were sintered at 900 °C yielding pellets with a two-phase structure; Fe2O3 and Fe3BO6. The sintered pellets, connected as cathode, were then electro-deoxidized in molten CaCl2 or in CaCl2–NaCl eutectic, against a graphite anode at 3.1 V. The electrolysis at 850 °C has successfully yielded a powder mixture of Fe and Fe2B. Sequence of changes during the electrolysis was followed by interrupted experiments conducted at 850 °C. This has shown that iron is extracted quite early during the electrolysis through the depletion of oxygen from the starting oxide; Fe2O3, forming the other iron oxides in the process. Boron follows a more complicated route. Fe3BO6, the initial boron-bearing phase, was depleted in the early stages due to its reaction with molten salt. This gave rise to the formation of calcium borate. Boron was extracted from calcium borate in later stages of electrolysis, which appeared to have reacted in situ with the iron forming compound Fe2B.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Direct synthesis of metals and compounds from their oxides are of considerable interest [1]. A major route in achieving this is the so-called electro-reduction process, first proposed in 1990s by Fray [2]. This is a reduction process in the solid sate where the oxides connected as cathode are electrolyzed in a molten salt against a graphite anode. In this process, according to Chen et al. [3], the oxygen in the cathode is ionized, carried in the electrolyte, and discharged from the anode in the form of CO or CO2 leaving behind the metallic constituents. According to Suzuki et al. [4], this is not a direct process but involves calciothermic reduction of the oxides, i.e., CaO acts as intermediary in the deoxidation process.
Since electro-deoxidation is a solid-state process, the preparation of oxide preform as cathode material has attracted considerable attention [5–8]. In the synthesis of pure metals, the interest is limited to ensure sufficient mechanical strength in the oxide preform so as to maintain its integrity during the electrolysis. An additional aspect of interest, as in the studies of Gordo et al. [5] and Centeno-Sanchez et al. [6], has been the control of porosity in the preform as a way of accelerating the reaction rate. In the case of alloys and compounds, the preform involves more than one oxide. The preform preparation then has a wider scope. The sintering of preform, in some cases, leads to the consolidation of the existing oxide phases, e.g., TiO2 and W2O3 as in the production of Ti-10 %W [7]. Mostly, however, the sintering leads to compounding between the oxides. An example for this is the formation of Tb3Fe5O12 and TbFeO3 from the mixture of Fe2O3 and Tb4O7 as reported by Qiu et al. [8].
As to how the reduction proceeds in the cathode during the electrolysis has also been of interest [9–11]. It has been found that the electro-deoxidation usually involves processes more complex than that of gradual removal of oxygen from phases as they exist in the preform. Studies by Jiang et al. [9] and Yan and Fray [11] have shown that intermediate phases, usually of perovskite type, do develop during the electrolysis of TiO2 and Nb2O5, which originate from the pellet–electrolyte interactions upon application of electrical potential. The occurrence of these intermediate phases has been clearly shown in partially reduced samples. A more systematic method, in this respect, has been the use of interrupted experiments used in the production of FeTi [12] where phase changes were followed sequentially during the course of electrolysis.
The present work deals with the synthesis of near eutectic Fe–4.6 wt% B alloy from the oxide mixture of Fe2O3 and B2O3 in molten salt. The system is of particular interest since Fe–B alloys of near eutectic compositions have potential as soft magnetic materials and more importantly, the mixture combines oxides of widely different melting points whose reduction behavior is also of interest.
Experimental
Starting materials were Fe2O3 and boric acid, both of technical grades. The powders were blended in proportions corresponding to the final metallic composition, i.e., Fe–4.6 wt% B. They were mixed in a Spex mill for 2 h with a ball-to-powder ratio of 1.5. The powder mixture was then cold compacted into a cylindrical pellet at a pressure of 110 MPa, yielding cylindrical pellets of 15 mm in diameter and 3–4 mm in height.
The pellets were sintered at 900 °C for selected periods of time under normal atmosphere in a vertical tube furnace. Heating rate was slow, typically 5 °C/min, so as to let the boric acid release its water, and convert to B2O3. This in-situ conversion was preferred since the water released could lead to generation of new and probably homogenously distributed pores in the structure.
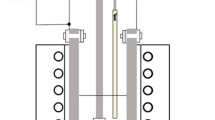
Electro-deoxidation experiments were carried out in an electrochemical cell shown schematically in Fig. 1. The cell comprises a stainless steel crucible holding the electrolyte, an anode (graphite), and two cathodes; one was the oxide pellet to be deoxidized and the other was an auxiliary electrode, i.e., stainless steel wire. The reactor was gas tight allowing electrolysis in an argon atmosphere (99.995% purity) maintained at a flow rate of 200 mL/min. The electro-deoxidation experiments were carried out at 850 °C in CaCl2 or 600 °C in eutectic mixture of CaCl2–NaCl. A total 1 kg of salts, technical grade, was used in each experiment.

Schematic representation of an electrolytic cell used in the deoxidation experiments
For each experiment, the fresh electrolyte was heated to electrolysis temperature slowly so as to reduce its moisture content. The electrolyte was further purified by pre-electrolysis using the graphite anode and the auxiliary cathode at a potential of 3.0 V for 8 h. After pre-electrolysis, the auxiliary cathode was lifted above the melt and the electrolysis was initiated by immersing the oxide pellet into the molten salt. The electrolysis was carried out at a constant potential of 3.1 V, up to 12 h. Here, the potential selected is safely below the decomposition voltage of the electrolyte both at 850 and 600 °C.
After electrolysis, the electrodes, i.e., pellet and the graphite anode, were removed from the electrolyte by lifting and positioning them above the melt. They were then allowed to cool to room temperature while maintaining the argon flow. Having cooled to room temperature, the reactor lid was opened and the pellet with its stainless steel connector was removed. The sample in the form of sponge like aggregate was washed in water and all undissolved material was collected as the reaction product.
Results and discussion
Compacts sintered at 900 °C for 1 h were in the form of an oxide mixture. Since B2O3 is a glassy phase, XRD pattern of this sample (not reproduced here) had Fe2O3 peaks only. Sintering for 2 h developed a new phase Fe3BO6, see Fig. 4a. The formation of this phase is thermodynamically favorable as indicated by the Fe2O3–B2O3 phase diagram [13, 14]. The oxide pellet sintered for 2 h, therefore, had a two-phase structure, i.e., Fe2O3 and Fe3BO6. The presence of Fe3BO6, instead of B2O3, is quite beneficial since the iron borate remains solid up to 900 °C. As a result, the sintering conditions were fixed for 2 h at 900 °C. SEM image of the sintered pellet is given in Fig. 2a. The pellets were quite porous with values in the range of 41–45%. These values were calculated from measurements on the pellets using the known densities of the constituent phases.

SEM images of a pellet derived from Fe2O3–10 wt% B2O3 compact via sintering at 900 °C for 2 h and b the pellet electro-deoxidized at 850 °C for 12 h in molten CaCl2 (3.1 V)
A typical current–time plot recorded during electro-deoxidation is given in Fig. 3. As seen in the figure, the current following a rapid rise to a peak value drops down sharply and then gradually decreases to smaller values finally settling at a residual current of about 0.5 A. This initial rise of current is usually attributed to the rapid surface metallization of the oxide pellet [15].

Current–time data collected during electro-deoxidation of the oxide pellet in CaCl2 at 850 °C (3.1 V)
After 12 h of electro-deoxidation, the pellet was fully reduced to a mixture of Fe and Fe2B, see Fig. 4d. There is no trace of oxides or elemental (crystalline) boron in the pattern. This shows that after 12 h of electro-deoxidation, the sample has been reduced fully to metallic form. Moreover, B has reacted with Fe to form the compound Fe2B. SEM image of the reduced sample is given in Fig. 2b. Based on data given in Fig. 3, the calculation yields a current efficiency of only 0.16 for this reduction. This is a rather low value but may be improved substantially if the residual current could be reduced to a fraction of its present value.

X-ray spectra of the oxide pellet sintered at 900 °C and electro deoxidized in CaCl2 at 850 °C (3.1 V). Spectra refer to pellets: a as-sintered condition; b, c, and d refer to sintered pellet electrolyzed for 30 min, 3 h, and 12 h, respectively
Following the successful electro-reduction of the oxides at 850 °C, an attempt has been made to carry out the electrolysis at a lower temperature. For this purpose, the oxide mixture, having been prepared in the same manner, was electrolyzed at 600 °C in a eutectic mixture of NaCl and CaCl2. XRD pattern of this sample is given in Fig. 5. Here, the pattern is dominated by a strong Fe phase, which is accompanied by some residual B-bearing oxides (see below). The pattern, as such, is intermediate between that of the starting oxides and that electrolyzed fully at 850 °C. Thus, it appears that electrolysis for duration of 12 h at 600 °C is not sufficient to reduce the oxides completely.

X-ray diffractogram of the oxide pellet electro-deoxidized in CaCl2–NaCl at 600 °C for 12 h (3.1 V)
The sequence of changes that brings the oxide mixture to the final metallic powder was examined by interrupted experiments. Since the kinetics was rather slow at 600 °C, interrupted experiments were conducted at 850 °C. Durations of 0.5 and 3 h were selected based on the current–time curve given in Fig. 3.
X-ray pattern after 3 h of electrolysis, shown in Fig. 4c, comprised Fe as the dominant phase. FeO was also present in the sample probably derived from partial deoxidation of Fe2O3. The sample did contain other phases that were difficult to identify. Calcium borate, Ca3B2O6, seemed to be present but could not be ascertained from the mentioned pattern.
So as to verify the presence of Ca3B2O6, a sample was prepared by mixing and compacting CaO and B2O3 in the molecular ratio of 3:1. The compact was then sintered for 4 h at 900 °C. The treatment yielded calcium borate, Ca3B2O6, as verified by its X-ray pattern given in Fig. 6. The peak positions given in this figure do match with most of the remaining peaks in Fig. 4c. Thus, it can be concluded that the chemical pathway of electro-deoxidation in the current system do involve the formation of calcium borate.

X-ray diffractogram of Ca3B2O6. The diffractogram refers to the pellet derived from the mixture of CaO and B2O3 in molar proportions of 3:1 sintered at 900 °C for 4 h
An interesting observation with regard to the 3-h deoxidized sample is that iron boron compounds have not yet formed in the sample. It may be worth mentioning that the same was also true for 12-h deoxidized sample at 600 °C, which in terms of its XRD pattern is quite similar to that of 3-h deoxidized sample. This shows that boron reduction occurs quite late in the process, in the time bracket between 3 and 12 h at 850 °C. Whether or not the current sample contains pure boron cannot be stated directly since boron can be in amorphous state and can therefore escape detection by X-rays. To check the stoichiometry of boron in the sample, a quantitative phase analysis was carried out via Rietveld refinement of the X-ray pattern. This gave values of 47, 33, 20 wt% for Fe, calcium borate, and FeO, respectively. This corresponds to Fe–4.7 wt% B, which is quite close to the targeted value (4.6 wt%). This implies that all boron is tied up in calcium borate in the sample and has not yet been reduced to elemental state.
X-ray diffractogram of the sample electro-deoxidized for 30 min show that iron is already a substantial phase. This verifies the observation made above that iron forms quite early in the process. The other phases present are FeO, Fe3O4, and possibly a Ca-bearing phase namely CaB2O4. Traces of Fe2O3 and Fe3BO6, which make up the sintered pellet could also be detected (compare Fig. 4a with b). It is interesting to note that within 30 min, there is a drastic diminution of the original Fe3BO6 phase.
Considering the sequence of changes from the oxide preform to the final product, the electro-deoxidation seems to follow quite a systematic route. At the beginning of the deoxidation, the initial two-phase structure was altered probably as a result of the pellet reacting with molten salt. More specifically, oxide pellet reacts with CaO, which normally presents in CaCl2 as a result of the reaction between CaCl2 and H2O during the drying process. The reaction leading to the consumption of Fe3BO6 probably occurs via
and/or
Calculation of standard Gibbs free energy using the thermodynamic data from Barin et al. [16] and data derived for Fe3BO6 phase from the phase diagram of B2O3–Fe2O3 [14] show that both of these reactions are quite favorable. Values of ΔG° are −68.1 kJ/mol of CaB2O4 for reaction (1) and −86.1 kJ/mol of Ca3B2O6 for reaction (2). Thus, CaO preferentially reacts with Fe3BO6 to form CaB2O4 or Ca3B2O6 within the oxide pellet. It is worth mentioning that similar reactions might be expected to occur between Fe2O3 and CaO forming compounds such as CaFe2O4, Ca2Fe2O5, and CaFe4O7. Values of ΔG° calculated for these reactions are −33, −61, and −29 kJ/mol in the respective order. The formation of these compounds, therefore, is not as favorable as those involving Fe3BO6, i.e., Eqs. (1) and (2).
It appears that the reduction process proceeds with Fe2O3 and calcium borate. Relevant reactions that may be involved in this process are given below. The standard reduction potentials, ΔE°, of these reactions, given by
have also been calculated using the thermodynamic data given in ref. [17]. Here, ΔG° is the standard Gibbs free energy change of the reaction (in J) at temperature T, n the number of electrons transferred in the balanced electrochemical reaction, and F is the Faraday’s constant (96,500 C/g equiv.). The values are calculated assuming that the materials are in pure state, reactions taking place at 850 °C, and the anode product being CO:
It should be noted that the reactions involving iron oxides, i.e., reactions (4)–(6), have positive reduction potentials implying that these reactions are spontaneous. This is consistent with the fact that iron is extracted quite early in the process. It should also be noted that the values of decomposition potentials for boron-bearing oxides are much more negative than those of iron oxide. This is particularly true for Ca3B2O6 where the value is ΔE° = −1.63 V. Thus, boron-bearing oxides are more difficult to reduce. This is probably the reason for the late appearance of boron-bearing iron compounds in the current experiments. It is worth noting that the value of decomposition voltage for Ca3B2O6 is even lower than those of B2O3 and Fe3BO6, compare reactions (7) and (8) with reaction (9). This implies that the reduction of boron is made more difficult by the formation of Ca3B2O6.
It is clear from above that, there are distinct differences as to how the reductions of iron and boron are achieved in the current oxide system. Iron seems to follow a direct route. Fe2O3 present in the pellet is reduced quite quickly through the depletion of its oxygen. Oxides lean in its oxygen content, e.g., Fe3O4 and FeO do form in the process as noted above in the partially deoxidized samples.
Boron, on the other hand, seems to follow a different route. Boron, initially present in Fe3BO6 in the sintered pellet, is not derived from this phase. Fe3BO6 phase is subject to early decomposition during the electrolysis. As discussed above, this probably occurs due to the reaction of Fe3BO6 with molten salt, more specifically with CaO, which, as stated above, is normally present in CaCl2. This leads to the formation of calcium borate as an intermediate phase. Thus, chemical pathway of boron reduction is not direct but proceeds from this intermediate phase. It should be mentioned that as far as the reduction of boron is concerned the current oxide system is quite similar to some of the other oxides, e.g., TiO2 and Nb2O5, which also form intermediate phases during their electro-deoxidation. It appears that the extraction of boron is delayed as a result and when extracted react with iron already reduced to elemental form, forming the compound Fe2B.
Results reported above show that the oxide mixture having a low melting point ingredient (B2O3) can be reduced quite successfully in molten CaCl2. The fact that B2O3 is liquid at the electrolysis temperature did not affect the deoxidation process; simply because on reacting with Fe2O3, it formed a high melting point oxide phase, Fe3BO6. The formation of this phase was therefore quite beneficial in this respect. As inferred from B2O3–Fe2O3 phase diagram [13, 14], Fe–B alloys up to 6 wt%B can be synthesized using the current approach without the risk of melting in the cathode, i.e., provided that the sintering temperature is high enough as in the current study, to form Fe3BO6.
Conclusions
In the current work, Fe–4.6 wt% B alloy was synthesized via electro-deoxidation of the mixed oxide precursor. The oxides, Fe2O3 and B2O3, mixed in suitable proportions were sintered at 900 °C yielding pellets with a two-phase structure, Fe2O3 and Fe3BO6. The sintered pellets connected as cathode were then electro-deoxidized in molten CaCl2 or in CaCl2–NaCl eutectic against graphite anode at 3.1 V. The study has shown the following:
-
1.
the electrolysis of the mixed oxide pellet at 850 °C for duration 12 h has successfully yielded the target composition in the form of a powder mixture of Fe and Fe2B.
The sequence of changes that occur in the pellet during the electrolysis at 850 °C was studied using the interrupted experiments. This has further shown the following:
-
2.
the reduction of iron oxide occurs quite early in the process through the depletion of oxygen from the starting oxide Fe2O3, forming other iron oxides and finally iron in the process;
-
3.
boron initially contained in Fe3BO6 has followed a different route. The reduction occurs from calcium borate, an intermediate phase formed after the depletion of Fe3BO6. Boron is, therefore, extracted late in the process and it seems to react with iron, forming the compound Fe2B.
References
Fray DJ, Chen GZ (2004) Mater Sci Technol 20:295
Fray DJ (2001) JOM 53:26
Chen GZ, Fray DJ, Farthing TW (2000) Nature 407:361
Suzuki RO, Tatemoto K, Kitagawa H (2004) J Alloys Compd 385:173
Gordo E, Chen GZ, Fray DJ (2004) Electrochim Acta 49:2195
Centeno-Sanchez RL, Fray DJ, Chen GZ (2007) J Mater Sci 42:7494. doi:https://doi.org/10.1007/s10853-007-1588-8
Dring K, Bhagat R, Jackson M, Dashwood R, Inman D (2006) J Alloys Compd 419:103
Qiu G, Wang D, Ma M, Jin X, Chen GZ (2006) J Electroanal Chem 589:139
Jiang K, Hu X, Ma M, Wang D, Qiu G, Jin X, Chen GZ (2006) Angew Chem 45:428
Wang SL, Xue Y, Sun H (2006) J Electroanal Chem 595:109
Yan XY, Fray DJ (2005) J Electrochem Soc 152:E308
Tan S, Örs T, Aydinol MK, Öztürk T, Karakaya I (2009) J Alloys Compd 475:368. doi:https://doi.org/10.1016/j.jallcom.2008.07.018
Makram H, Touron L, Loriers J (1972) J Cryst Growth 13–14:585
Cam I, Timuçin M (2004) In: Proceedings of the 2nd international boron symposium, Chamber of Mining Engineers of Turkey, Eskisehir, 2004, p 195
Tripathy PK, Gauthier M, Fray DJ (2007) Metall Mater Trans B 38:893
Barin I, Knacke O, Kubaschewski O (1973) Thermochemical properties of inorganic substances, 2nd edn. Springer, Berlin
Bale CW, Chartrand P, Degterov SA, Eriksson G, Hack K, Ben Mahfoud R, Melançon J, Pelton AD, Petersen S (2002) Calphad 26:189
Acknowledgement
This work is supported by Scientific and Technological Council of Turkey (TUBITAK MAG 105M352).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at https://doi.org/10.1007/s10853-009-3595-4
Rights and permissions
About this article
Cite this article
Örs, T., Tan, S., Öztürk, T. et al. Synthesis of Fe–4.6 wt% B alloy via electro-deoxidation of mixed oxides. J Mater Sci 44, 3514–3519 (2009). https://doi.org/10.1007/s10853-009-3474-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-009-3474-z