
Abstract
Iron oxide colloids were prepared by coprecipitation of Fe(II) and Fe(III) salts in alkaline media and stabilized by perchloric acid, oleic acid, or poly(acrylic acid). In an attempt to obtain magnetic polymer microspheres differing in size, dispersion polymerization of glycidyl methacrylate (GMA) in ethanol containing HClO4-stabilized magnetite, dispersion copolymerization of GMA and 2-hydroxyethyl methacrylate (HEMA) in toluene/2-methylpropan-1-ol mixture in the presence of oleic acid-coated magnetite, and inverse suspension copolymerization of N-isopropylacrylamide (NIPAAm) and N,N′-methylenebisacrylamide (MBAAm) in cyclohexane in the presence of poly(acrylic acid)-coated maghemite were compared. The microspheres were characterized by morphology, size, polydispersity, and some magnetic properties.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Magnetic particles find a wide range of in vivo and in vitro biomedical applications, such as cell separation and purification [1, 2], MRI [3, 4], magnetic hyperthermia [5, 6] and drug targeting. Magnetic separation can advantageously replace classical separation techniques, such as centrifugation, filtration, or chromatography. A variety of materials were described for the formulation and preparation of magnetic polymer microspheres.
The presence of functional groups on the microsphere surface that are suitable for covalent attachment of target biomolecules or other compounds is an essential condition for immunomagnetic applications. At the same time, functional monomers, in particular hydrophilic and water-soluble ones, present in the polymerization feed, can considerably affect the mechanism of formation of polymer particles and their magnetic properties. Therefore we have selected two reactive methacrylate-based and one thermoresponsive polymer differing in hydrophilicity as a basis for the preparation of magnetic polymer microspheres. The aim is to find the best possible support suitable for the separation of biomolecules. Methods of preparation of the microspheres (dispersion vs. inverse suspension polymerization) were compared, as well as the properties of the resulting microspheres in terms of morphology, size, polydispersity, and some magnetic properties. Magnetic polymer microspheres were synthesized from glycidyl methacrylate (GMA), GMA, and 2-hydroxyethyl methacrylate (HEMA), and N-isopropylacrylamide (NIPAAm). The advantage of PGMA consists in the presence of oxirane groups which can be easily modified, e.g., hydrolyzed to vicinal diols [7], transformed to amines, or oxidized to aldehyde groups [8]; also −SO3 −, –N⊕R3, and chelating and other functional groups can be introduced [9]. Moreover, PGMA microspheres, which are less hydrophilic than PHEMA, are well dispersible in water. PHEMA is well known for its nontoxicity and is thus widely used in biomedical applications. Nonionic poly(N-isopropylacrylamide) (PNIPAAm) is an important representative of temperature-sensitive gels [10, 11]. One of the reasons why it has been widely studied is that PNIPAAm can shrink and/or swell in water in response to temperature changes and even undergo a large volume change at a temperature of ∼32 °C, which is called the volume-phase transition temperature (VPTT). The temperature about 32 °C is close to human temperature (37 °C) and hence such PNIPAAm hydrogels have been evaluated in temperature-controlled drug delivery systems [12–14] and separation processes [15]. In these systems, the substrates in PNIPAAm hydrogels are squeezed out in the target tissue in the body by shrinking the gels in response to temperature. Moreover, thermosensitive materials may yield surfaces that control biological interactions, such as bioadhesion [16, 17].
Experimental
Materials
2-Hydroxyethyl methacrylate (HEMA), glycidyl methacrylate (GMA), N-isopropylacrylamide (NIPAAm), N,N′-methylenebisacrylamide (MBAAm), poly(acrylic acid) (Mw = 2,000), 50 wt% solution in water, were from Aldrich (Milwaukee, WI, USA). Ammonium persulfate (APS) was from Lachema (Brno, Czech Republic), 2,2′-azobisisobutyronitrile (AIBN) was obtained from Fluka (Buchs, Switzerland) and crystallized from ethanol. Poly(N-vinylpyrrolidone) (PVP K30; Mw = 40,000) from Fluka, and Kraton G 1650 (poly(styrene-block-hydrogenated butadiene-block-styrene); Mw = 74,000, Mn = 70,000; PSt/PBu = 29/71 w/w) from Shell (Houston, USA) were the stabilizers. Cellulose acetate butyrate CAB 381-20 was from Eastman Chemical Company (Kingsport, USA); Span 80 (sorbitan monooleate) and other solvents and reagents were from Aldrich.
Synthesis of colloid iron oxide nanoparticles
Primary magnetite nanoparticles were prepared by coprecipitation of FeCl2 and FeCl3 (1:2 molar ratio) upon addition of NH4OH. In a typical reaction, 80 mL of 0.5 M NH4OH was added to 80 mL of 0.2 M FeCl3 under 5 min sonication at 50% power (W 385 Ultrasonic Sonicator; Cole-Palmer Instruments, USA), which was followed by the addition of 40 mL of 0.2 M FeCl2. The mixture was poured into 240 mL of 0.5 M NH4OH under stirring (2,000 rpm, anchor-type stirrer) resulting in the precipitation.
To get an acid colloid, the Fe3O4 precipitate was stirred with 40 ml of aqueous 2 M perchloric acid (HClO4) and isolated by centrifugation for 40 min at 2,500 rpm. Its peptization was accomplished by adding ethanol (20 g) and sonicating for 10 min.
Alternatively, 5 ml of oleic acid was added to the above-described primary precipitate and the reaction mixture heated for 5 h at 90 °C. After cooling to room temperature, the nanoparticles were washed with water for 4 days (200 mL of water three times a day) and dried at 80 °C/13 Pa.
To obtain poly(acrylic acid)-coated maghemite nanoparticles, the magnetite precipitate was first left to grow for 30 min, and washed with water (5 × 20 mL) until peptization was achieved. The magnetite colloid was then oxidized with 10 ml of 5 wt% NaOCl aqueous solution under sonication followed by the addition of 13 mL of 0.1 M sodium citrate again under 5 min sonication. The product was washed as described above, and sonicated for 5 min. Five milliliters of 0.4 wt% poly(acrylic acid) was added to 5 mL of the colloid containing 33 mg γ-Fe2O3/mL. Finally, the volume of the dispersion was reduced by evaporation to 3 mL.
Preparation of magnetic PGMA microspheres by dispersion polymerization
About 2.4 g of PVP K30 stabilizer was dissolved in 68 mL of an ethanolic acid ferrofluid containing 0.6 g of Fe3O4. To this a solution of 0.03 g of APS initiator in 12 mL of GMA was added and the mixture bubbled with nitrogen for 10 min. The polymerization was run in a 100 mL reactor equipped with an anchor-type stirrer at 70 °C with stirring (500 rpm) for 17 h. After completion, some blank (nonmagnetic) microspheres were easily removed by replacing the supernatant during purification by repeated magnetic separation from ethanol. While magnetic particles concentrated at the bottom, the upper layer was decanted. The procedure was repeated (up to 20 times) until excessive PVP was removed and there was no more solid in the supernatant. To remove excessive magnetite (not embedded inside the polymer matrix), particles were washed with 2 M HCl for 24 h and water; they were then transferred to ethanol and finally dried from ether.
Preparation of magnetic P(HEMA-co-GMA) microspheres by dispersion polymerization
About 42.2 g of toluene was added to 1 g of oleic acid-coated Fe3O4 and sonicated for 20 min at 50% power; spontaneous redispersion proceeded, creating a homogeneous and black dispersion. Then, a solution of 3.2 g CAB in 25.8 g of 2-methylpropan-1-ol was added, and after sonication, the flask containing this dispersion was mounted to a reactor. 6 g of HEMA, 6 g of GMA, and 0.24 g of dibenzoyl peroxide were added, and the solution was deoxygenated by purging with nitrogen for 10 min. The reaction was run at 70 °C with stirring (500 rpm) for 17 h. The resulting particles were purified by repeated magnetic separation from a toluene/2-methylpropan-1-ol mixture (with gradually decreasing content of 2-methylpropan-1-ol) and decantation until excess CAB was removed and there was no more solid in the supernatant solution. Then, the product was dried from toluene.
Preparation of magnetic P(NIPAAm-co-MBAAm) particles by inverse suspension polymerization
The continuous phase was prepared by dissolving 3 g of Kraton G 1650 and 0.075 g of AIBN in 75 mL of cyclohexane in 100 mL reactor and 4 g of Span 80 was added. The dispersed phase consisted of 1.5 g of the above poly(acrylic acid)-coated γ-Fe2O3 ferrofluid with 2 g of NIPAAm and 0.1 g of MBAAm dissolved for 5 min at 70 °C. The dispersed phase was mixed with the continuous phase under sonication at laboratory temperature, the mixture was purged with nitrogen, and the polymerization was performed for 8 h at 70 °C. The resulting particles were washed 10 times with hexane (50 mL each) and dried in air.
Characterization
Optical microscope Meopta (Přerov, Czech Republic) equipped with a Minolta Dimage A1 camera and scanning electron microscope (SEM; JEOL JSM 6400) were used for viewing the microspheres. For the SEM viewing, a drop of dilute microsphere dispersion in water was spread on the glass surface and dried in a dust-free environment at room temperature. The dried sample was then sputtered under vacuum with a thin layer (~4 nm) of platinum. Transmission electron microscope (TEM) micrographs of iron oxide nanoparticles were obtained with JEOL JEM 200 CX. Particle size and distribution were determined by measuring at least 300 particles for each sample using software Atlas (Tescan, Brno, Czech Republic). Two types of the mean particle size were calculated: number-average (Dn) and weight-average (Dw), (Dn = ∑Di/N, Dw = ∑Di4/∑D 3 i , Di is the particle diameter, N is the number of particles). Particle size distribution was characterized by polydispersity index (PDI) calculated as a ratio of weight- to number-average particle diameter.
The amount of iron in the microspheres was analyzed by AAS (Perkin-Elmer 3110) of an extract from the sample obtained with dilute HCl (1:1) at 80 °C for 1 h. The phase composition of the magnetic iron oxide nanoparticles/polymer composite microspheres and the crystallite size of magnetic particles were determined by X-ray powder diffraction using a Bruker D8 diffractometer (CuKα radiation, Sol-X energy dispersive detector). X-ray diffraction patterns were analyzed by the Rietveld method using the FULLPROF program (Version 2.20-Sep2002-LLB JRC). The crystallite size delimits the XRD coherence length and thus contributes to peak width. The Thompson-Cox-Hastings pseudo-Voigt profile was used to resolve instrumental, strain and size contributions to peak broadening. Instrumental resolution was determined by measuring strain-free tungsten powder with crystallite size 9.4 μm. Magnetic properties were measured with a SQUID magnetometer MPMS5 (Quantum Design, San Diego, USA) at 295 K at the fields up to 1.5 kA m−1.
Results and discussion
Synthesis of particles
Magnetite was synthesized in a two-step process by alkaline coprecipitation of iron(II) and iron(III) precursors in aqueous solution followed by the addition of various stabilizers. The resulting nanoparticles were viewed by TEM; their mean size polydispersity are given in Table 1. Magnetite nanoparticles stabilized electrostatically by the addition of HClO4 had a moderate tendency to form chains (Fig. 1a). The particles were smaller than 10 nm in diameter (Table 1). In contrast, the use of the hydrophobic stabilizer, oleic acid, led to individual, well-separated magnetite nanoparticles of a spherical shape and rather narrow size distribution with the mean diameter ca. 10 nm (Fig. 1b). Finally, maghemite nanoparticles obtained by the oxidation of primary magnetite and coated with poly(acrylic acid) (PAA) were slightly larger (Fig. 1c) compared with the former. They had a tendency to aggregate probably due to the sticky character of PAA. Nevertheless, the described effects of the stabilizer on the morphology and size of iron oxide seemed to be only minor. However, the surface coating affected the selection of the polymerization medium.

TEM micrograph of (a) HClO4-stabilized Fe3O4 nanoparticles, (b) oleic acid-coated Fe3O4 nanoparticles, and (c) PAA-coated γ-Fe2O3
Depending on the selected monomer, different solvents had to be used as a continuous medium for the heterogeneous polymerization to obtain magnetic polymer particles. While ethanol was suitable for the dispersion polymerization of GMA, copolymerization of HEMA and GMA was run in a mixture of toluene/2-methylpropan-1-ol in order to fulfill the essential condition required by dispersion polymerization—the solubility of the monomer but insolubility of the polymer in the polymerization medium. In contrast, suspension polymerization of NIPAAm, which is a water-soluble monomer, was run in cyclohexane, in which the monomer does not dissolve. The choice of the polymerization medium then dictated both the selection of a suitable ferrofluid added in the polymerization feed and the selection of the stabilizer. HClO4-stabilized magnetite nanoparticles were therefore found suitable for the PVP-stabilized dispersion polymerization of GMA in ethanol, oleic acid-coated magnetite colloid was stable in CAB-stabilized dispersion copolymerization of HEMA and GMA in a toluene/2-methylpropan-1-ol mixture, while poly(acrylic acid)-coated maghemite could be used for Kraton G 1650/Span 80-stabilized inverse suspension copolymerization of NIPAAm and MBAAm in cyclohexane. These three different methods for preparation of magnetic polymer microspheres were compared in terms of their effect on the properties of the resulting product. Some of the properties of the microspheres are summarized in Table 1. Values of the saturated magnetizations give an evidence of the expected increase of the magnetization with increasing content of the ferrimagnetic iron oxide.
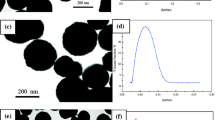
The magnetic PGMA microspheres obtained by dispersion polymerization of GMA initiated with ammonium persulfate (APS) in ethanol containing the poly(vinylpyrrolidone) stabilizer in the presence of HClO4-treated Fe3O4 were rather small, with the mean size 150 nm, but polydisperse (Fig. 2a). Because homogeneous nucleation accompanied the formation of magnetic microspheres, white nonmagnetic particles were also produced in the polymerization. However, they were removed during magnetic separation. As a result, the remaining magnetic PGMA microspheres contained a relatively large amount of iron (Table 1).

SEM micrograph of (a) magnetic PGMA microspheres, (b) magnetic P(HEMA-co-GMA) microspheres and (c) optical micrograph of dry magnetic P(NIPAAm-co-MBAAm) microspheres
In an attempt to narrow the particle size distribution, HEMA was dispersion copolymerized with GMA in toluene/2-methylpropan-1-ol. Magnetic microspheres larger by an order of magnitude than the PGMA microspheres were thus obtained by dibenzoyl peroxide (BPO)-initiated and cellulose acetate butyrate (CAB)-stabilized copolymerization of HEMA and GMA in toluene/2-methylpropan-1-ol in the presence of oleic acid-coated Fe3O4. The main advantage of the procedures consists in that it yielded almost monodisperse microspheres with PDI = 1.05 (Fig. 2b). The P(HEMA-co-GMA) microspheres contained more than 5 wt% of iron.
Finally, the largest microspheres in this study were obtained by AIBN-initiated and Kraton G 1650/Span 80-stabilized inverse suspension copolymerization of NIPAAm and MBAAm in cyclohexane in the presence of poly(acrylic acid)-coated γ-Fe2O3. The resulting P(NIPAAm-co-5%MBAAm) microspheres containing ca. 7 wt% of iron were slightly polydisperse (PDI = 1.16) with the average diameter of 54 μm. They were accompanied by tiny nonmagnetic particles (Fig. 2c).
Phase analysis
X-ray powder analysis of the microspheres documented the presence of a polymer shell. The X-ray diffraction pattern confirmed the presence of the iron oxide core spinel phase, either magnetite or maghemite, in all the polymer microspheres (Fig. 3). Because both the phases have nearly identical lattice constants, it is difficult to decide which particular phase exists in an individual polymer sample. No additional investigation like measurements of Mössbauer spectra carried out in our previous study [18] was performed, and therefore the phase type present in the particular microspheres could be deduced only from the preparation method. Iron oxide crystallite sizes (coherent length) determined by X-ray analysis agreed with the results of TEM (Table 1).

X-ray powder analysis of (a) magnetic PGMA microspheres, (b) magnetic P(HEMA-co-GMA) microspheres, and (c) magnetic P(NIPAAm-co-MBAAm) microspheres. v—nonmagnetic microspheres; w—magnetic microspheres containing iron oxide spinels; vertical bars—Fe3O4 or γ-Fe2O3 standard
Magnetic characteristics
The obtained microspheres were indeed magnetic as they were rapidly collected with a magnet. Magnetization curves of the magnetic PGMA, P(HEMA-co-GMA), and P(NIPAAm-co-MBAAm), microspheres are shown in Fig. 4. An expected gradual increase in the saturated magnetization with increasing content of the iron spinel phase in the microspheres was observed in accordance with the results of chemical analysis (Table 1).

Magnetization curves of magnetic PGMA (u), P(HEMA-co-GMA) (v), and P(NIPAAm-co-MBAAm) microspheres (w)
Conclusions
In an attempt to obtain magnetic polymer microspheres differing in size, three heterogeneous polymerization techniques in the presence of iron oxide were selected: dispersion polymerization of GMA in ethanol yielding polydisperse submicrometer particles, dispersion copolymerization of HEMA and GMA in a toluene/2-methylpropan-1-ol mixture providing monodisperse micron-size microspheres, and finally inverse suspension copolymerization of NIPAAm and MBAAm producing microspheres in the range of tens of micrometers. While the former microspheres possess functional groups for immobilizing various compounds, the latter microspheres can respond to temperature. The hybrid microspheres thus show a potential in magnetic and biomedical applications.
References
Radisic M, Iyer RK, Murthy SK (2006) Int J Nanomed 1:3. doi:https://doi.org/10.2147/nano.2006.1.1.3
Xia N, Hunt TP, Mayers BT, Alsberg E, Whitesides GM, Westervelt RM et al (2006) Biomed Microdevices 8:299. doi:https://doi.org/10.1007/s10544-006-0033-0
Bulte JWM, Kraitchman DL (2004) NMR Biomed 17:484. doi:https://doi.org/10.1002/nbm.924
Lewin M, Carlesso N, Tung Ch H, Tang XW, Cory D, Scadden DT et al (2000) Nat Biotechnol 18:410. doi:https://doi.org/10.1038/74464
Fukumori Y, Ichikawa H (2006) Adv Powder Technol 17:1. doi:https://doi.org/10.1163/156855206775123494
Wust P, Gneveckow U, Johannsen M, Bohmer D, Henkel T, Kahmann F et al (2006) Int J Hyperthermia 22:673. doi:https://doi.org/10.1080/02656730601106037
Tennikova TB, Horák D, Švec F, Kolář J, Čoupek J, Trushin SV et al (1988) J Chromatogr A 435:357. doi:https://doi.org/10.1016/S0021-9673(01)82196-8
Lenfeld J, Švec F, Kálal J (1986) Acta Polym 37:377. doi:https://doi.org/10.1002/actp.1986.010370612
Kenawy ER, Abdel-Hay FI, El-Shanshoury AR, El Neseny MH (1998) J Control Release 50:145. doi:https://doi.org/10.1016/S0168-3659(97)00126-0
Heskins M, Guillet EJ (1968) J Macromol Sci Part A 2:1441
Shibayama M, Isono K, Okabe S, Karino T, Nagao M (2004) Macromolecules 37:2909. doi:https://doi.org/10.1021/ma0359685
Yoshida R, Sakai K, Okano T, Sakurai Y (1993) Adv Drug Deliv Rev 11:85. doi:https://doi.org/10.1016/0169-409X(93)90028-3
Castro Lopez V, Hadgraft J, Snowden MJ (2005) Int J Pharm 292:137. doi:https://doi.org/10.1016/j.ijpharm.2004.11.040
Lee CF, Wen CJ, Lin CL, Chiu WY (2004) J Polym Sci Part Polym Chem 42:3029. doi:https://doi.org/10.1002/pola.20085
Seida Y, Nakano Y (1996) J Chem Eng Jpn 29:767. doi:https://doi.org/10.1252/jcej.29.767
Lahann J, Mitragotri S, Tran TH, Kaido H, Sundaram J, Choi IS et al (2003) Science 299:371. doi:https://doi.org/10.1126/science.1078933
Ohya S, Kidoaki S, Matsuda T (2005) Biomaterials 26:3105. doi:https://doi.org/10.1016/j.biomaterials.2004.08.006
Pollert E, Knížek K, Maryško M, Závěta K, Lančok A, Boháček J et al (2006) J Magn Magn Mater 306:241. doi:https://doi.org/10.1016/j.jmmm.2006.03.069
Acknowledgements
Financial support of the Grant Agency of AS CR (grant KAN200200651) and the Academy of Sciences of the Czech Republic (grant 1QS100100553) is gratefully acknowledged. The authors thank Mrs. J. Hromádková of the Institute for electron microscopy measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Horák, D., Pollert, E. & Macková, H. Properties of magnetic poly(glycidyl methacrylate) and poly(N-isopropylacrylamide) microspheres. J Mater Sci 43, 5845–5850 (2008). https://doi.org/10.1007/s10853-008-2836-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-008-2836-2