
Abstract
Liquid polycarbosilane (LPCS) with a highly branched structure was characterized by fourier-transform infrared spectrometry (FT-IR) and 1H, 13C, 29Si nuclear magnetic resonance spectrometry (NMR). The LPCS was then cured and pyrolysized up to 1,600 °C under flowing argon. The structural evolution process was studied by thermogravimetric analysis and differential scanning calorimetry (TG-DSC), FT-IR, and X-ray diffraction (XRD). Hydrosilylation, dehydrocoupling, and polymerization cross-linking reactions between Si–H and C=C groups occurred at low temperatures, which mainly accounted for the high ceramic yield (70%) up to 1,400 °C. The organic groups gradually decomposed and the structure rearranged at high temperatures. The FT-IR analysis revealed that Si–CH2–Si chains, the backbone of original polymer, can be retained up to 1,200 °C. At temperatures higher than 1,200 °C, the Si–CH2–Si chains broke down and crystalline SiC began to form. The final crystalline products were β-SiC and a small amount of carbon.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Silicon-based ceramics have attracted great interest for high-temperature applications due to their high mechanical strength, hardness, and corrosion resistance at elevated temperatures [1]. SiC/Si3N4 ceramics can be synthesized by thermal decomposition of polymer precursors [2]. This chemical-to-ceramics route possesses many unique advantages over conventional powder-based ceramic processing, such as fabricating unconventional structures including fibers [3], ceramic matrix composites [4], and ceramic micro-electro-mechanical system (MEMS) [5]. The micro-structure of final ceramics can be controlled by varying the compositions and structures of polymer precursors. Due to no oxide additives evolving in the process, polymer-derived ceramics exhibit excellent high-temperature properties. Polymer-derived SiCN and SiBCN ceramics have shown unique thermal stability [6], creep resistance [7, 8], and high-temperature electric properties [9]. SiAlCN ceramics thus obtained demonstrated extremely high oxidation resistance [10, 11] and corrosion resistance [12, 13], suitable for combustion environment applications.
Polycarbosilanes have attracted particular interest because they are important precursors to synthesize SiC-based ceramics. Although some attempts to prepare silicon-based materials from polymer precursors in the 1960s, the potential of preceramic compounds in the area of ceramics was not recognized until the pioneer work of Yajima [14], who prepared precursor-derived SiC ceramic fibers. After that, a large number of attempts to synthesize effective silicon carbide ceramics have been made from precursors other than Poly(dimethylsiloxane) PDMS [15–17]. Most efforts have been focused on polycarbosilanes, i.e., polymer with Si–C bonds in their backbones. Several routes have been used to obtain such polymers, which were systematically summarized in reference [18]. Today, the research in polycarbosilanes focuses not only on the polymer precursor itself, but also on the polymer cross-linking process, polymer–ceramic conversion, and the thermostability of pyrolyzed ceramics [19–21]. Polycarbosilanes were also applied to ceramic matrix composites [22] and foams [23], in addition to non-oxide ceramic fibers.
In this paper, we report the study of the conversion of a new liquid polycarbosilane to ceramics. The highly branched structure was confirmed by FT-IR and NMR spectra. The curing, pyrolysis, and crystallization processes were characterized by FT-IR and XRD. It was interesting to notice that the backbone structure of the as-received polymer could be retained up to 1,200 °C. The crystallization behavior also depends on the broken backbone chains. The results indicated that the structure of the final ceramics can be manipulated by designing the initial polymer precursors. Hence, the properties of ceramics thus obtained can be controlled intentionally.
Experimental procedure
A liquid polycarbosilane (LPCS) with highly branched structure was synthesized in the Laboratory of Advanced Materials at Xiamen University. Briefly, LPCS was synthesized by Grignard coupling of chloromethylmethyldichlorosilane (Cl2MeSiCH2Cl), (chloromethyl)trichlorosilane (Cl3SiCH2Cl), and allyl chloride (ClCH2CH=CH2), followed by the reduction with lithium aluminum hydride (LiAlH4) [24]. The structure of the as-received LPCS was characterized by FT-IR and NMR spectroscopy. FT-IR spectra were obtained by placing the liquid on NaCl plates using a Nicolet Avator 360 Spectrometer (Wisconsin, USA). NMR experiments were carried out on a Bruker AV300 MHz spectrometer (Germany) operating at 300.13 MHz for 1H, 75.46 MHz for 13C (1H decoupling), and 59.63 MHz for 29Si (1H decoupling, delay time 30 s). The specimen used for NMR was dissolved in CDCl3 solution. The 1H, 13C, and 29Si chemical shifts were all referred to (trimethylsilyl)silane (TMS).
The cross-linking process was carried out at 170 °C for 6 h under argon protection. The samples for the polymer-to-ceramics conversion study were treated at different temperatures in argon for 1 h. The structures of the resultant solids were characterized by FT-IR, using pellets made from the mixture of the solid powders and dried KBr powders. Thermal analysis for the cross-linking and conversion process was carried out on a thermal gravimetric analysis-differential scanning calorimetry (TGA-DSC) (Netzsch STA 409C, Germany) in argon gas with a ramping rate of 10 °C/min.
The crystallization behavior of the pyrolysized amorphous ceramics was studied by X-ray diffraction (XRD) and FT-IR. XRD was carried out using a Rigaku D/max-2400 diffractometer (Tokyo, Japan) with copper Kα radiation. Data were digitally recorded in a continuous scan in the range of angle (2θ) from 10° to 75° with a scanning rate of 0.08°/s. FT-IR was performed just as described above for solid samples.
Results and discussion
Characterization of liquid polycarbosilane
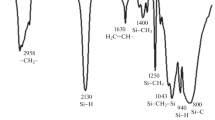
To identify the functional units present, the synthesized liquid polycarbosilanes are characterized by FT-IR (Fig. 1). The peaks at around 1,050 cm−1 (CH2 bending in Si–CH2–Si) and 2,920 cm−1(C–H stretching in Si–CH2–) indicate the existence of a Si–CH2–Si chain, the backbone of polymer. The bands attributed to Si–CH3 are Si–CH3 stretching at 1,250 cm−1 and C–H stretching in Si–CH3 at 2,950 cm−1. A strong band at 2,120 cm−1 is assigned to Si–H. The C–H vibration in CH=CH2 at 3,073 cm−1 and the C=C stretching at 1,630 cm−1 indicate that the vinylsilyl group (CH2=CH–) exists in this LPCS [25–27].

FT-IR analysis of synthesized liquid polycarbosilanes
The structure of liquid polycarbosilane is further confirmed by NMR spectra (Fig. 2). The peaks in 1H NMR spectrum of the LPCS (Fig. 2a) are overlapped and broadened, as expected on the basis of the highly branched structure of LPCS. The signals centered at −0.2 ppm are the overlap of SiCH2Si and SiCH3 [28]. The peak at 0.15 ppm is attributed to SiCH2– [29]. The SiCH2Si and SiCH2– should be generated by Grignard coupling reaction to form the backbone of the polymer. The multiplet at around 1.85 ppm is due to the methylene protons of the residual chloromethyl functionalities not completely reduced by LiAlH4 and the methylene protons derived from THF [29]. As reported previously [29], chlorosilanes underwent a side reaction with ether, resulting in the incorporation of methylene from THF into the polymer chain. The three peaks ranged from 3.3 ppm to 4.4 ppm match the values for the SiH, SiH2, and SiH3 groups [30]. The two multiples at 4.77 and 5.76 ppm are assigned, respectively, to the protons of CH=CH*2 and CH*=CH2 [31], indicating that the allyl groups have been introduced to the side chain of the polymer.

NMR analysis of synthesized liquid polycarbosilanes (a) 1H NMR (b) 13C{1H} NMR (c) 29Si{1H} NMR
The 13C NMR spectrum of LPCS (Fig. 2b) further clarifies the polymer structure. Due to the numerous environments around silicon element, the complex multiplets from −15 ppm to +8 ppm can all be assigned to Si–CH2–Si chain [29, 31]. The peak centered at 27 ppm is attributed to residual SiCH2C1 functionality that is not completely reduced by the LiAlH4 [32]. Peaks at 44.83, 62.40, and 70.88 ppm are consistent with products resulting from the cleavage of the THF solvent [32]. Signals at 114.02 and 133.84 ppm, attributed to carbons from C*H2=CH and CH2=C*H, also validate the existence of the allyl groups [31].
The 29Si NMR spectrum (Fig. 2c) shows a number of peaks, indicating that silicon connects with different functionalities. The peak centered at 2 ppm is attributed to (CH2)4Si units. The groups of peaks between −8 ppm and −15 ppm and several resolved singlets ranging from −53 ppm to −67 ppm originate from the silicon in (CH2)3SiH and (CH2)SiH3 units in the polymer, respectively [29]. Those between −26 and −39 should be generated from (CH2)2SiH2 units in LPCS [29, 32]. The peak at around −110 ppm is due to the existence of sample tube. The complexity of 29Si NMR spectrum of the polymer further reflects a branched structure of LPCS.
From the above analysis, it is believed that the polymer precursor has a highly branched structure with a Si–CH2–Si chain (The structure is shown below). The highly branched structure is critical to a high final ceramic yield [33]. The introduction of vinyl function group into the polymer will enhance the cross-linking of polymer, which will result in a higher ceramic yield. The structural effect on the ceramic yield will be discussed in detail in another paper.

Cross-linking of liquid polycarbosilane
Figure 3 compares the FT-IR spectrum of the as-received liquid PCS with the spectrum of the cured one. The absorption peaks related to Si–H (2,130, 900 cm−1) and SiCH3 (1,250, 2,950, 2,900, 1,400, and 1,355 cm−1) greatly decreased in intensity after curing. The C–H stretching in –CH=CH2 (3,073 cm−1) almost disappears and the intensity of the absorption band attributed to C=C (1,630 cm−1) is reduced a lot, which indicates that the CH2=CH– functionality evolves in the cross-linking reaction. It is believed that hydrosilylation and dehydrocoupling reactions occurred during the cross-linking process, as shown below:

Comparison of FT-IR spectra for (a) as-received liquid polycarbosilanes and (b) cured polycarbosilanes at 170 °C
Other reactions may also occur as follows:
The study of Choong Kwet Yive et al. [34] indicated that the cross-linking activity can be arranged in the following order: hydrosilylation>dehydrocoupling>vinyl group polymerization. Hence, the reaction (1) and (2) should be the major reactions during cross-linking. It was further confirmed that the ratio of Si–H to Si–CH3 decreased from 10.01 for the as-received PCS to 7.19 for the cured one.
Active units, such as Si–H and –CH=CH2, will enhance the cross-linking degree, thus improve the final ceramic yield. It is confirmed by the fact that the weight content of Si–CH=CH2 increasing from 3% to 9% leads to an improvement of ceramic yield from 60% to 71%. However, the vinyl function group will result in excessive free carbon in the final ceramics.
Pyrolysis of liquid polycarbosilane
The pyrolysis behavior of the cross-linked solids is investigated by TGA-DSC. The polymer exhibits three-stage weight loss at around 240 °C, 380 °C, and 550 °C, as indicated in TGA-DSC curve (Fig. 4). The corresponding weight losses are 2 wt%(25–290 °C), 24 wt%(290–490 °C), and 9 wt% (290–780 °C), with an overall value of 35 wt% at 800 °C.

TGA-DSC analysis of cured polycarbosilanes
The polymer–ceramic conversion process of the solidified PCS is also studied by using FT-IR (Fig. 5). The spectrum of the sample heat-treated at 300 °C is similar to that of the one cured at 170 °C, except that the intensity of the peaks related to the Si–H group (2,130, 900 cm−1) is reduced, which can be explained by the hydrosilylation and dehydrocoupling reactions for the further cross-linking. Hence, the weight loss for the first stage is due to evaporation of the reaction product H2 [34] and some small molecules. From the spectrum at 400 °C, it can be seen that the spectrum is also similar to the one treated at the lower temperatures. However, the intensities of Si–H group (2,130, 900 cm−1) are further reduced. The functionality of Si–CH3 (1,250, 2,950, 2,900, 1,400, and 1,355 cm−1) and CH2=CH– (3,073, 1,630 cm−1) is also decreased. It is believed that the polymer continues cross-linking. Additional reactions of (3) and (4) also happen during this temperature range [33]. Gaseous products such as CH4 and H2 will evaporate out, contributing to the weight loss in the second stage. Nevertheless, the 24 wt% weight loss in this period cannot totally come from the gaseous products. The low weight molecules that require further cross-linking will escape out at high temperatures, which should also account for the weight loss. This is confirmed by the fact that a sample treated at 280 °C for 10 h in argon has a 10 wt% weight loss less than the one without being treated.

FT-IR spectra of polycarbosilanes heat-treated at (a) 300 °C, (b) 400 °C, (c) 600 °C, (d) 900 °C, (e) 1,200 °C, (f) 1,400 °C, and (g) 1,600 °C
For the spectrum at 600 °C, the intensities of peaks for Si–H and Si–CH3– are all greatly reduced. This should be due to the decomposition of organic side groups [25, 35]. The evolution in this temperature range contributes greatly to the removal of excess carbon. It can also be noticed in the FT-IR spectrum that the C=C peak at 1,630 cm−1 becomes stronger, which is due to the formation of free carbon in the sample. The XRD pattern (Fig. 6) also confirms the existence of carbon in the sample treated at 600 °C.

XRD patterns of polycarbosilanes heat-treated at (a) 600 °C, (b) 900 °C, (c) 1,000 °C, (d) 1,200 °C, (e) 1,400 °C, and (f) 1,600 °C
The spectrum of sample treated at 900 °C becomes featureless. The broad peak at around 800 cm−1 is attributed to SiC4 functionality. A very small C=C peak (1,630 cm−1) also appears. Other functionalities such as Si–CH3, Si–H, even SiCH2Si are no longer present. It is believed that the conversion from polymer to ceramics is complete at around 900 °C. From the TGA-DSC result, no obvious weight loss is observed at around 900 °C and an exothermic peak appears, which indicates that a structural rearrangement happens at this temperature.
Crystallization behavior
XRD and FT-IR are used to investigate the crystallization behavior of pyrolyzed samples. After the conversion from polymer to ceramic is complete at 900 °C, the material is amorphous except for a small amount of crystalline free carbon inside. Further treating the sample at 1,000 °C causes a very broad peak to appear at 36°. Although the crystallization is incomplete, it indicates that a high local order of SiC4 exists in the sample. From the FT-IR spectrum (Fig. 5), it is interesting to notice that Si-CH2-Si functionality (1,020, 1,046 cm−1) appears again. At temperature of 900 °C, the samples should be highly disordered. The featured peaks of Si-CH2-Si and SiC4 overlap with each other to form a broad band. At 1,000 °C, SiC4 becomes more ordered and its peak will be distinguished from the Si–CH2–Si peak. The XRD and FT-IR features for the samples treated at 1,200 °C are similar to those at 1,000 °C, except that the intensity of Si-CH2-Si is reduced. It indicates that the Si–CH2–Si gradually breaks down at high temperatures to give way to SiC4. The peaks of β-SiC shown in XRD patterns are very broad at the temperature of 1,200 °C, indicating that the particle size of SiC is very small (∼7 nm). It is believed that such a small size comes directly from a decomposition of Si–CH2–Si. Si–CH2–Si functionality totally disappears at the temperature of 1,400 °C, while large amount of β-SiC are formed at this temperature. Heat-treating the samples at the temperature of 1,600 °C, causes the peaks from β-SiC to become sharper and the crystal size also to grow larger (∼33 nm) as expected.
Based on the above FT-IR and XRD analysis, we can describe the structure of the amorphous ceramics derived from liquid polycarbosilane as follows: the major building units are SiC4 and Si–CH2–Si that comes from the chain of original polymer. These units are connected through C–C or C=C to form a 3-D network.
Conclusions
In this paper, the structure of a new liquid polycarbosilane is characterized by NMR and FT-IR. The results indicate the polymer is highly branched with a Si–CH2–Si chain.
The cross-linking mechanism is studied. It is believed that hydrosilylation and dehydrocoupling are the main reactions during cross-linking. The pyrolysis and crystallization process of the cured polymer is studied by FT-IR and XRD. The Si–CH2–Si chain of polymer can be kept to high temperatures and strongly affect the crystallization temperature. This research also demonstrates that the final structure of ceramics can be manipulated by controlling the original polymer structure.
References
Goto Y, Thomas G (1995) J Mater Sci 30:2194
Kroke E, Li YL, Konetschny C et al (2000) Mater Sci Eng R 26:97
Yajima S, Hasegawa Y, Okamura K et al (1978) Nature 273:525
Zbigniew SR (2001) J Am Ceram Soc 84:2235
Liew L, Zhang W, An L, Shah S et al (2001) Am Ceram Soc Bull 80:25
Riedel R, Kienzle A, Dressler W et al (1996) Nature 382:796
An L, Riedel R, Konetachny C et al (1998) J Am Ceram Soc 81:1349
Riedel R, Ruwisch LM, An L et al (1998) J Am Ceram Soc 81:3341
Ramakrishnan PA, Wang YT, Balzar D et al (2001) Appl Phys Lett 78:3076
Wang Y, Fan Y, Zhang L et al (2005) J Am Ceram Soc 88:3075
Wang Y, Fan Y, Zhang L et al (2006) Scripta Mater 55:295
Wang Y, Fei W, An L (2006) J Am Ceram Soc 89:1079
Wang Y, Fei W, Fan Y et al (2006) J Mater Res 21:1625
Yajima S, Hayashi J, Omori M et al (1976) Nature 261:683
Takeda M, Imai Y, Ichikawa H et al (1992) Ceram Engi Sic Prog 13:209
Kriner WA (1964) J Org Chem 29:1601
Schilling CL, Wesson JP, Williams TC (1983) Am Ceram Soc Bull 62:912
Birot M, Pillot JP, Dunogues J (1995) Chem Rev 95:1443
Narisawa M, Kitano S, Idesaki A et al (1998) J Mater Sci 33:2663
Bouillon E, Langlais F, Pailler R et al (1991) J Mater Sci 26:1333
Janakiraman N, Weinmann M, Schuhmacher J et al (2002) J Am Ceram Soc 85:1807
Ly HQ, Taylor R, Day RJ (2001) J Mater Sci 36:4027
Fitzgerald TJ, Mortensen A (1995) J Mater Sci 30:1025
Huang TH, Yu ZJ, He XM et al (2007) Chin Chem Lett 18:754
Bouillon E, Langlais F, Pailler R et al (1991) J Mater Sci 26:1333
Hasegawa Y, Okamura K (1986) J Mater Sci 21:321
LY HQ, Taylor R, Day RJ et al (2001) J Mater Sci 36:4037
Froehling PE (1993) J Inorg Organomet Polym 3:251
Whitmarsh CK, Interrante LV (1991) Organometallics 10:1336
Matthews S, Edirisinghe MJ, Folkes MJ (1999) Ceram Int 25:49
Rushkin IL, Shen Q, Lehman SE et al (1997) Macromolecules 30:3141
Michalczyk MJ, Davidson F (1994) Monatshefte für Chemie 125:895
Gonon MF, Hampshire S, Dissod JP et al (1995) J Eur Ceram Soc 15:683
Choong Kwet Yive NS, Corriu RJP, Leclerq D et al (1992) Chem Mater 4:141
Hasegawa Y, Iimura M, Yajima S (1980) J Mater Sci 15:720
Acknowledgements
The project was supported by the Natural Science Foundation of Fujian Province of China (No. E0510002).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, H., Zhang, L., Cheng, L. et al. Polymer–ceramic conversion of a highly branched liquid polycarbosilane for SiC-based ceramics. J Mater Sci 43, 2806–2811 (2008). https://doi.org/10.1007/s10853-008-2539-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-008-2539-8