
Abstract
Surfactant-mediated intercalation of aliphatic carboxylic acids into a commercial layered double hydroxide (LDH) with approximate composition \( [{\hbox{Mg}}_{{0.689}} {\hbox{Al}}_{{0.311}} ({\hbox{OH}})_{2} ](\text{CO} _{3} )_{{0.156}} \, \cdot \,0.5{\hbox{H}}_{{\hbox{2}}} {\hbox{O}} \)] was explored. The reaction was conducted at elevated temperatures with the LDH powder as a suspension in a stearic acid oil-in-water emulsion. The acidic fatty acid, e.g., stearic acid, reacts with the basic carbonate anions, CO2 is released and the fatty acid is intercalated as a bilayer. High-concentration anionic or nonionic surfactants, i.e., sodium dodecylsulfate or Tween 60, aid the intercalation process by emulsifying the molten acid and dispersing the hydrotalcite particles. X-ray diffraction, thermal analysis, and infrared spectroscopy confirmed that a bilayer-intercalated hydrotalcite was formed and that the surfactant is not co-intercalated. The method is convenient, economical, and environmentally friendly: It employs the readily available carbonate form as starting reagent; water is used as medium rather than organic solvents; low reaction temperatures suffice, i.e., calcinations of the clay are superfluous and there is no need for working under a CO2-free atmosphere.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrotalcite is a natural anionic clay mineral [Mg6Al2(OH)16CO3·4H2O]. Layered double hydroxides (LDH) are synthetic analogues with the general composition \( [{\hbox{M}}^{{{\hbox{II}}}}_{{1 - x}} {\hbox{M}}^{{{\hbox{III}}}}_{x} ({\hbox{OH}})_{2} ]^{{x + }} A^{{y - }}_{{x/y}} \, \cdot \,z{\hbox{H}}_{{\hbox{2}}} {\hbox{O}} \) with MII = Mg, Zn, Fe, Co, Ni, Cu; MIII = Al, Fe, Cr; A represents a suitable anion and \( 1\,\,\, \le \,\,5x\,\, < \,\,2 \) [1–3]. These compounds have a brucite-like [Mg(OH)2] stacked sheet structure in which the cations are octahedrally coordinated by six oxygen atoms as hydroxides. The sheets have a net positive charge owing to the partial replacement of Mg2+ with Al3+ ions. The interlayer contains water and charge balancing anions, e.g., CO 2−3 , Cl−, or stearate, etc. [4]. The corresponding compounds are herein referred to as LDH-CO3, LDH-Cl, or LDH-St. The d-spacing of the (003) planes in brucite and the CO 2−3 containing LDH structures are 0.47 nm and 0.76 nm, respectively. Layered double hydroxides are stable in the range 3 < pH < 12 [4].
Self-assembly is the process whereby small pre-existing subunits spontaneously organize themselves into an ordered state or structural arrangement. The formation of lamellar micelles by surfactants molecules in solution is a typical example [5]. Interactions applicable for self-assembly include electrostatic attractions, hydrogen bonding, and hydrophobic interactions among others [6]. Intercalation is a form of self-assembly that involves the trapping of guest molecules between layers in a crystal lattice. Intercalation is the reversible insertion of a mobile guest species into a crystalline host lattice during which the structural integrity of the latter is formally conserved [7]. In principle, layered host lattices can adapt to the geometry of the inserted guest species by adjustment of the interlayer separation. Chemical bonding is not a prerequisite but the process may involve ion exchange. Layered hosts such as LDH feature strong intralayer covalent bonding and weaker interlayer interactions. Linear molecules with appropriate functional groups may self-assemble into monolayers or bilayers between the sheets of LDH [7].
Carlino [8] reviewed the methods for preparing LDH intercalated with carboxylic acids and their salts. Intercalated LDH can be prepared by direct synthesis methods [9]; e.g., hydrothermal crystallization of gels formed by the coprecipitation of the MII and MIII hydroxides in the presence of the required organic anion [10–12]. This method works well with terephthalate and arene sulfonates [11]. However, with alkyl carboxylates and α,ω-dicarboxylates it yields products with diffuse or even amorphous X-ray patterns [10, 11]. Furthermore, the products are usually contaminated with the precursor anions from the mixed salt solutions [13, 14]. An ingenious solution to this problem was devised [15]. LDH-CO3 is dissolved in excess aqueous carboxylic acid and the required pure product obtained by precipitation by addition of the mixture to a basic solution. Presumably this approach is limited to water-soluble acids, e.g., hydroxycarboxylic acids such as citric, tartaric, and malic acid.
Indirect methods utilize suitable LDH precursors prepared by direct synthesis. Three main indirect techniques were identified [9]: (i) direct anion exchange; (ii) LDH reconstruction from a layered double oxide form obtained by calcinations of a suitable precursor; and (iii) anion replacement by elimination of the precursor interlamellar species.
Direct anion exchange
In the direct ion exchange method [4, 10, 16–19], the carboxylic acid is incorporated into LDH-A by direct contact with a suitably concentrated aqueous or non-aqueous solution of either (a) the desired carboxylic acid or (b) an appropriate carboxylic acid salt [8]. Owing to the tenacity by which carbonate is held, it is customary to use either A = chloride or nitrate.
Miyata and Kumura [4] pioneered direct ion exchange. They exchanged the interlamellar anions in Zn3Al-LDH with a series of α,ω-dicarboxylic acids. Meyn et al. [16] exchanged Mg3Al-LDH-NO3 with organic acid (sodium) salts dissolved in water. Borja and Dutta [17] intercalated lauric, myristic, and palmitic acid forms by treating LDH-Cl with ethanol solutions of the corresponding fatty acids. XRD evidence indicated incomplete reaction with some of the precursor material still present. The samples also retained significant amounts of ethanol as evidenced a large mass loss of ca. 30% upon heating to 90 °C. XRF analysis also showed evidence that chloride ion was retained. The Cl− anions in LDH-Cl were exchanged by treatment with aqueous solutions of sodium stearate under a nitrogen atmosphere [18, 19]. In both cases impure intercalates were formed that contained remnants of the initial anions.
Crepaldi et al. [9] describe an exchange method based on the formation and organic phase extraction of a salt between dodecylsulfate and a cationic surfactant.
Dimotakis and Pinnavaia [20] claim that LDH ion exchange reactions are topotactic. If true, this implies that any layer stacking defects in the precursor will also appear in the pillared product.
The LDH reconstruction method
The LDH reconstruction method comprises a hydrothermal reconstitution of calcined LDH carbonates in the presence of the desired anion in carbonate-free water [21–24]. Owing to strong electrostatic and hydrogen-bonding interactions, carbonate anions do not readily ion exchange. Calcining a suitable precursor, e.g., LDH-CO3 at 400–500 °C produces a dehydroxylated and decarbonated layered double oxide (LDO) form. From this the original clay can be reconstructed, in an intercalated form, by treatment with an aqueous solution of the required anion. The mechanism is believed to entail the fast rehydration of the oxide with intercalation of OH− anions, followed by a slow anion exchange of the latter with other anions [24]. The formation of the pure hydroxide (LDH-OH) form requires reconstruction in pure water and total exclusion of CO2, e.g., a nitrogen atmosphere [20].
However, when applied to organic anions, mixed phases may be produced [23], and it is difficult to avoid formation of carbonate forms. An improvement to this route was proposed by reacting carboxylic acids with the LDH-OH form in the presence of glycerol as “swelling agent” [20]. Intercalation of carboxylic acids and other anions are facilitated by the presence of glycerol [20, 25]. The latter intercalates, causing the LDH basal spacing to increase. It is believed that the glycerol in the interlayer facilitates exchange with other anions by decreasing the strength of electrostatic and hydrogen-bonding forces acting on the carbonate ions.
LDH has also been reconstructed by the reaction of LDH-OH with acid chlorides [26]. The LDH-CO3 was calcined and reconstituted to the LDH-OH by treatment with water and subsequently dehydrated in vacuo. This product was then suspended in organic solvents such as ether or acetonitrile containing acid chlorides. Morioka et al. [26] claim that their procedure led to esterification of the layer hydroxyl groups.
Anion exchange by elimination of the precursor interlamellar species
Thermal intercalation takes place when the LDH-CO3 is brought into direct contact with pure molten organic acids, e.g., sebacic acid [27, 28]. However, despite long reaction times of up to 10 h, the conversion was incomplete resulting in polyphasic reaction products.
Prevot et al. [29] compared the coprecipitation, anion exchange, and reconstruction methods for the synthesis of 2:1 Zn,Al-LDH-succinate and tartrate. They found that ion exchange is a suitable method for both acids. Coprecipitation did not lead to high crystallinity for the LDH-tartrate. The reconstruction method failed entirely. No LDH was formed owing to the complexing power of the hydroxyacid. Prevot et al. [30] found that oxalate can be intercalated as such or as an anionic aluminum complex.
Higher fatty acids are usually intercalated as bimolecular layers [17]. Depending on factors such as the temperature and the relative amount of stearate employed, either a monolayer or a bilayer of stearate is intercalated. The interlayer distance is about 3.2 nm for the monolayer and ca. 5.1 nm for the bilayer [18, 31]. Since stearate intercalates to form a close-packed bilayer structure, the amount intercalated is independent of the anion exchange capacity (AEC) [19].
Kanoh et al. [18] intercalated sodium stearate into 2.25:1 Mg-Al-LDH-Cl. The clay was suspended in aqueous solution of sodium stearate for 1 day at different temperatures. They showed that stearate self-assembly in LDH reversibly changes between mono- and bilayer structures depending on the intercalation temperature: For 1 AEC sodium stearate, the lower temperatures, e.g., 5 °C, favored monolayer intercalation while at higher temperatures, e.g., 70 °C, bilayer was preferentially formed. However, the bilayer also formed at the lower temperature provided an excess of sodium stearate is supplied. Their powder X-ray diffraction results show that highest purity is attained at ca. 3 AEC. They found that the sodium salts of octanoic and decanoic acids did not intercalate. Kanoh et al. [18] attribute bilayer formation in terms of hydrophobic interactions. They justify this as follows: If methanol instead of water is used, temperature and sodium stearate concentration has no effect. Only 1 AEC is intercalated in monolayer format.
Itoh et al. [19] intercalated sodium stearate into 3:1 or 4:1 Mg,Al-LDH-Cl. The clay was suspended in aqueous solution of sodium stearate for 1 day at different temperatures dependent on the aliphatic acid in question. The intercalates contained stearate as counter ion with the excess in the form of sodium stearate and free stearic acid. They attribute the presence of the free acid to contamination with atmospheric CO2.
The distinctive properties of modified layered double hydroxides permit a wide range of uses including polymer additives [32], precursors for catalysts [33, 34], and magnetic materials [32]. Their generally non-toxic nature and membrane-like structure can be harnessed to protect, carry, deliver, and controllably release pharmaceuticals or genes [31, 33]. Nanocomposites can be prepared by exfoliation within polymer matrices [33, 35–39].
The intercalation procedures described above all suffer from some of the following problems: (i) Poorly crystallized products; (ii) contamination with other anions and solvents, and (iii) mixed phases containing one or more of the following: Precursor material owing to incomplete reaction or ion exchange and reaction with CO2 or incomplete decomposition of the carbonate. Owing to the wide-ranging utility of LDH intercalates, it is of interest to develop environmentally friendly and energy-efficient methods of intercalation that yield purer products. This work reports on a facile one-pot method for the intercalation of long-chain carboxylic acids into LDH-CO3. In essence it is an extension of the Carlino melt method [27, 28, 40] involving surfactants as intercalation aids. Sodium dodecylsulfate (SDS) was found to be a suitable surfactant for the purpose.
It was previously reported that anionic surfactants such as sodium dodecylsulfate (SDS) and sodium dodecylbenzenesulfonate (SDBS) adsorb on the LDH-CO3 crystal surfaces rendering them hydrophobic [41–43]. Surfactants do not intercalate when the LDH contains the non-exchangeable carbonate anion [43]. However, anionic surfactants do intercalate via ion exchange when the anion is nitrate [2] or chloride ion [44, 45].
Experimental
Materials
LDH-CO3 (Hydrotalcite Grade HT 325) was supplied by Chamotte Holdings. It contained silica and magnesium carbonate as minor impurities. Distilled water was employed in all experiments. Other chemicals used were: acetic acid (98%, Saarchem); acetone (99% C.P. Radchem Laboratory Suppliers); 25% aqueous ammonia solution (Promark Chemicals); dodecanoic acid (lauric acid, Croda Chemicals); docosanoic acid (behenic acid, Fluka); ethanol (96% rectified, Dana Chemicals and 99.9% absolute ethanol, A.R., Radchem Laboratory Suppliers); octanoic acid (caprylic acid, Croda Chemicals); polyoxyethylene (20) sorbitan monostearate (Tween 60, Sigma); potassium bromide (Uvasol KBr, Merck); sodium dodecylsulfate (SDS, 98%, Fluka), and stearic acid (St, AR, 65–90%, Bio-Zone Chemicals).
Sample preparation
Typical intercalation experiments were conducted according to the following procedure: 20 g LDH-CO3 \( [{\hbox{Mg}}_{{0.689}} {\hbox{Al}}_{{0.311}} ({\hbox{OH}})_{2} ](\text{CO} _{3} )_{{0.156}} \, \cdot \,0.5{\hbox{H}}_{{\hbox{2}}} {\hbox{O}} \)]) (0.0852 mol Al), 33.3 g stearic acid (0.117 mol), and 40 g SDS surfactant (0.139 mol) were suspended in 1.5 L distilled water. The mixture was heated to- and then maintained at the required reaction temperature, e.g., 80 °C, for 9 h and then allowed to cool down overnight. NH4OH was added to maintain pH > 10. This cycle was repeated four times. Two additional portions of stearic acid (33.3 g) were added after the first two cycles, so that the overall total was the required amount (100 g). In the last cycle the mixture was simply allowed to stir for 9 h without acid addition. The mixture was allowed to cool down slowly to ambient. The solids were recovered by centrifugation, washed once with distilled water, four times with ethanol, and thereafter once with acetone. After each washing the solids were separated from the liquid by centrifugation. The product was allowed to dry at room temperature. In some instances the product was further purified by Soxhlet extraction with ethanol to remove excess stearic acid.
Similar procedures were followed using octanoic (caprylic), dodecanoic (lauric), and behenic acids. Different intercalation temperatures were explored using lauric acid and stearic acid. For the latter, lower acid to clay ratios corresponding to ca. 1, 2, and 4 AEC were also investigated. The non-ionic surfactant polyoxyethylene (20) sorbitan monostearate (Tween 60) was explored as a replacement for the anionic SDS.
The standard intercalation experiment described above was repeated without adding a carboxylic acid but using double the amount of SDS. Fourier-Transform Infrared (FTIR), Thermogravimetry (TG), and X-Ray Diffraction (XRD) data for the product obtained showed that sodium dodecylsulfate on its own did not intercalate into LDH-CO3. Monolayer SDS intercalated LDH was prepared according to a method to be reported elsewhere [46].
Characterization
The precursor hydrotalcite was characterized as follows: Particle size distribution and BET surface area were determined using a Malvern Mastersizer Hydro 2000MY instrument and a Micromeritics Flowsorb II 2300 instrument, respectively. Purity was checked using a wavelength-dispersive X-ray Fluorescence (XRF) spectrometer (ARL 9400 XP + XRF). The powders were ground in a tungsten carbide milling vessel and roasted at 1000 °C for loss on ignition (LOI) determination. Samples were pelletized using ethyl cellulose as binder. An XRF analysis was also conducted on the residue obtained by ashing LDH-stearate (SDS) samples at 700 °C.
Small amounts of the powder product or LDH-CO3 precursor were placed onto carbon tape on a metal sample holder. Excess powder was removed using a compressed air blast. Samples were coated five times with gold using Scanning Electron Microscope (SEM) autocoating unit E5200 (Polaron equipment LTD) under argon gas. Gold-coated particles were viewed on a JEOL 840 SEM scanning electron microscope under low magnification.
A Mettler Toledo A851 simultaneous TGA/SDTA machine was used for thermal and gravimetric analysis. Powder samples (ca. 10 mg) were placed in open 70 μL alumina pan and heated from 25 °C to 700 °C at a scan rate of 10 °C/min in air.
Differential Scanning Calorimetry (DSC) data was collected on a DSC Q100 TA instrument. 5–10 mg samples were placed in an alumina pan and heated from −20 to 150 °C at a rate of 10 °C/min and a N2 flow rate of 50 mL/min.
Infrared spectra were recorded on a Perkin Elmer Spectrum RX I FT-IR system using the KBr method. The pellets contained approximately 2 mg of sample and 100 mg of KBr. Data obtained from 32 scans recorded at a resolution of 2 cm−1 were averaged and background-corrected using a pure KBr pellet.
Phase identification was carried out by XRD analysis on a PANalytical X-pert Pro powder diffractometer with variable divergence- and receiving slits and an X’celerator detector using Fe-filtered Co K-alpha radiation (0.17901 nm) at the University of Pretoria. X’Pert High Score Plus software was used for phase identification. Temperature-resolved XRD traces were obtained using an Anton Paar HTK 16 heating chamber with a Pt-heating strip. Scans were measured between 2θ = 1° to 40° in a temperature range of 25–150 °C in intervals of 25 °C with a waiting time of 1 min and measurement time of 6 min per scan. Si (Aldrich 99% pure) was added to the samples so that the data could be corrected for sample displacement using X’Pert Highscore plus software. It is presented as variable slit data as that allows for better data visualization.
Results and discussion
Unless stated otherwise, LDH-stearate (SDS) specifically refers to the product obtained reacting the equivalent of 4.12 times the AEC of stearic acid with LDH-CO3 at 80 °C for 24 h and SDS as surfactant.
Table 1 gives the normalized chemical analysis of the hydrotalcite grade HT 325 that was used as raw material. It also lists the chemical composition of an ashed LDH-stearate sample. The reported composition of the LDH-CO3 is consistent with an Mg:Al ratio of 2.21:1 (mol basis). The theoretical AEC of this LDH-CO3 is 402 meq/100 g. The ashed LDH-stearate (SDS) sample suggests instead an Mg:Al ratio of 1.93:1 (mol basis) and indicates the presence of almost one sodium atom for every two aluminum atoms.
The Malvern particle size analysis of the LDH-CO3 revealed a bimodal particle size distribution with d(0.1) = 1.0 μm; d(0.5) = 3.5 μm, and d(0.99) = 260 μm. The measured BET surface area was 12.9 m2/g.
Figure 1 shows that the LDH-CO3 (Hydrotalcite HT 325) consisted of numerous smaller crystals inter-grown in a “sand-rose” arrangement [47]. After reaction with stearic acid this structure was replaced by low-aspect ratio flakes that were significantly larger, as much as 20 μm across. The change in crystal size and habit indicates that the intercalation was accompanied by recrystallization, i.e., that the stearate intercalation process did not proceed in the topotactic manner suggested [20]. This is not surprising considering that stearate monolayers provide a template for the growth of Mg,Al-LDH [48].

SEM pictures showing: (a) The “sand rose” structure of the LDH-CO3 (Hydrotalcite HT 325) crystals. (b) The flake-like habit of LDH-stearate crystals prepared using SDS surfactant. (c) Delamination of the product in (b) after extraction with ethanol. In both cases the LDH-stearate flakes are significantly larger that the precursor crystals. (d) LDH-stearate crystals obtained with Tween 60 as surfactant
Figure 2 compares the thermo-gravimetric curves obtained for stearic acid, LDH-CO3, and LDH-stearate. The derivative curve (DTG) for LDH-CO3 is shown amplified by a factor of three. Mass loss of LDH-CO3 proceeds stepwise with three distinct but overlapping peaks in the DTG trace. These events are commonly attributed to the loss of interlayer water, dehydroxylation and a combination dehydroxylation–decarbonation reaction respectively [10, 21, 49, 50]:


Thermogravimetric mass loss (TG) and derivative curves for stearic acid, LDH-CO3 (HT 325), and LDH-stearate showing that the stearic acid has reacted or intercalated. The DTG curve for LDH-CO3 is shown amplified by a factor of three
For LDH-CO3 with x = 0.311, the expected and experimentally observed values for the TGA residues after the final degradation step are 56.3% and 58.3% (at T = 700 °C), respectively.
The sharp peak centered at 100 °C in the LDH-stearate corresponds to the loss of ca. 4% by mass of interlayer water in LDH-stearate. The other LDH-carboxylates showed similar mass losses at ca. 100 °C. The DTG peak at ca. 225 °C in the stearic acid trace is attributed to the vaporization of the carboxylic acid and its degradation products. In LDH-stearate this peak is almost entirely suppressed and new decomposition peaks appear in the DTG trace near 380 and 440 °C. These observations are consistent with intercalated stearate.
Figure 3 compares the FTIR spectra of LDH-stearate with those for stearic acid, LDH-CO3, and LDH-SDS in the wave number range 1150–1750 cm−1. In this range the LDH-SDS spectrum features a strong peak at 1210 cm−1 characteristic of the sulfate functional group. The carboxylate (COO−) asymmetric stretching vibrations bands at 1540, 1554, and 1588 cm−1 for the LDH-stearate correspond closely with those observed for meristate intercalated Mg3Al-LDH [17]. The ν(C=O) stretching vibration at 1702 cm−1, observed for stearic acid, is absent but the small carbonate peak at 1367 cm−1 indicates the presence of unreacted LDH-CO3 as an impurity. The absence of a sulfate peak at 1210 cm−1 [24], in this and the other LDH-carboxylate FTIR spectra, indicates that SDS did not co-intercalate with octanoic, dodecanoic, stearic, or behenic acids. The preferentially intercalation of long-chain fatty acids at the expense of SDS is attributed to a greater affinity of LDH for carboxylate.

FTIR spectra for the precursors LDH-CO3 and stearic acid and the products LDH-stearate and LDH-SDS
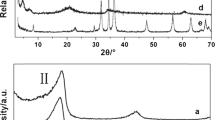
Figure 4 shows the X-ray diffractograms for stearic acid (99%), LDH-CO3 and LDH-stearate prepared with SDS as surfactant at an intercalation temperature of 80 °C. The reflections at 0.76 nm (2θ = 13.49°) and 0.38 nm (2θ = 27.21°) for LDH-CO3 are also present in the LDH-stearate confirming that the former was present as an impurity. The reflections at 5.04 nm (2θ = 2.04°), 2.56 nm (2θ = 4.01°), 1.72 nm (2θ = 5.97°), 1.036 nm (2θ = 9.91°), and 0.406 nm (2θ = 25.45°) are consistent with bilayer intercalated LDH-stearate [17, 19]. The linear alkyl substituents of the intercalated carboxylates assume an extended chain conformation [18]. It is further assumed that the slant angle of the alkyl chains is length-independent and that the methylene bond length equals to 0.127 nm. The d-spacing of a bilayer intercalated structure can then be expressed by the following correction to the equation presented [2, 8, 27]:
Here n represents the number of carbon atoms in the carboxylic acid; α is the slant angle of the carboxylate chain from the normal to the LDH layer plane (see Fig. 5); d o is the distance between the terminal (ionized) carboxyl group and the center plane of the brucite-like sheets; and d 2 is the distance between the two facing terminal methyl groups in the bilayer structure. A least-square fit of the present data shown in Fig. 5 yields:
This slope of 0.240 nm per –CH2– unit is intermediate to values of 0.235 and 0.2454 previously reported [17, 19]. According to Eq. 1, these slopes correspond to slant angles of 19.3°, 22.3°, and 15°, respectively. Figure 5 also plots the d-spacing values reported by Itoh et al. [19]. The literature values are slightly higher than those currently determined. The difference might be due to the presence of ethanol [17] or additional sodium ions [19] in the clay galleries.

X-ray diffractograms for stearic acid (99%), LDH-CO3, and LDH-stearate prepared with sodium dodecylsulfate (SDS) as surfactant at a reaction temperature of 80 °C and Soxhlet extracted with ethanol

Effect of carboxylic acid chain length on the d-spacing of intercalates prepared by the SDS-mediated intercalation method (●). The d-spacing for LDH-CO3 (■) and LDH-SDS (Δ) are also shown. The d-values reported by Borja and Dutta [17] (○) and Itoh et al. [19] (□) are slightly higher than the present ones
The aluminum ions are most probably randomly distributed within the Mg-Al-(OH) x sheets. However, the idealized regular arrangement shown in Fig. 6 aids the estimation of the projected surface area per formula weight of the LDH. The projected Mg-Al-(OH)x sheet area per LDH formula weight ([Mg2+αAl1−α(OH)6] (CO3)(1−α)/2·1.5H2O) is given by:
The lattice parameter equals a = 0.305 for the present LDH-CO3 [3]. This gives A LDH = 0.242 nm2.

(a) Idealized regular arrangement of aluminum (dark circles) and magnesium (open circles) in the metal hydroxide sheets of [Mg2+αAl1−α(OH)6] (CO3)(1−α)/2·1.5H2O with \( \alpha \cong 0 .\) The lattice parameter equals ca. a = 0.305 [3]. (b) Hexagonal close packing of the stearate chains
The cross-sectional area per stearic acid chain, assuming a hexagonal close packing, is given by:
The reflection 2θ = 25.422° (CoKα) indicates a layer spacing of t = 0.407 nm. Thus A chain = 0.191 nm2, i.e., the expected value for close packing of extended alkane chains. Noting the double layer intercalation and incorporating a correction for the slant angle, leads to the following estimate for the maximum intercalation level:
In the present case the slant angle is estimated at α = 19.1°. This yields a limit of 2.39 mol carboxylate/LDH for close-packed carboxylate chains.
The effect of stoichiometry
The degree of carboxylic acid intercalation was quantified using TG data obtained in air as follows: The interlayer water content was estimated from the mass loss recorded up to 148 °C. The clay content was calculated from the high temperature (700 °C) residue on the assumption that it only contains the metal oxides MgO, Al2O3, and sodium carbonate. These values were used to estimate the effective clay content. The carboxylic acid content was then calculated by difference. Figure 7 illustrates the effect of reagent stoichiometry. It compares the present data, obtained at an intercalation temperature of 80 °C, to values reported by Itoh et al. [19]. The apparent degree of stearate intercalation lies slightly above the theoretical curve for the product obtained at a feed composition of stearate/LDH = 4.12, and purified by Soxhlet extraction with neat ethanol. In contrast, the literature data [19] fall slightly below the expected curve.

Effect of reagent stoichiometry on the degree of intercalation stearic acid at (80 °C). The triangles indicate values for sodium stearate as measured by Itoh et al. [19]
The effect of reaction temperature
The effect of temperature on the degree of intercalation for stearic and lauric acids is shown in Fig. 8. Kanoh et al. [18] and Itoh et al. [19] report that intercalation of the water-soluble sodium stearate proceeded at temperatures as low as 5 °C. With the present procedure only incomplete intercalation of the acids was observed at temperatures below ca. 60 and 53 °C, respectively. The apparent degrees of intercalation exceeded the expected theoretical limit for intercalation temperatures above 60 °C. The reason for this is not currently understood. Kanoh et al. [18] state that, at low temperatures, monolayer intercalation of sodium stearate occurs whereas currently only bilayer intercalation of carboxylic acids was observed.

Effect of reaction temperature on the degree of intercalation of lauric and stearic acids
Differential scanning calorimetry (DSC)
Figure 9 shows the DSC melting endotherms for the pure and technical grade stearic acids, magnesium stearate, and the LDH-stearate (SDS). Table 2 compares the endotherm peak temperatures and the endotherm enthalpies. The technical grade stearic acid melts at a lower temperature and has a lower melting enthalpy than the 99% grade, as it also contains significant amounts of palmitic acid. The LDH-stearate was prepared by intercalating technical grade stearate. This compound shows two endotherm peaks. The one centered at ca. 107 °C corresponds to the dominant endothermic event. The main endotherm peak is positioned about 50 °C higher than in the parent stearic acid. This reflects the effect of the two-dimensional constraints imposed by the rigid inorganic sheets on the stearate bilayers. Magnesium stearate has the highest melting range. Its melting enthalpy is also larger than that observed for the stearic acids but it is lower than that for the LDH-stearate. In this compound all the stearic acid radicals are fully ionized and directly coordinated to magnesium atoms located in octahedral [51, 52]. By contrast, in LDH-stearate only a portion of the stearic acid molecules in the LDH galleries are expected to be ionized.

DSC melting endotherms for stearic acid (technical), stearic acid (99% pure) LDH-stearate (SDS), and magnesium stearate. The measured enthalpies were −158, −221, −241, and −173 kJ/kg, respectively
Hot stage microscopy
Figure 10 shows a DSC heating scan for LDH-stearate (Tween 60). This material also showed two melting endotherms. The second endotherm peak is more pronounced than that for the LDH-stearate (SDS) material in Fig. 9. Also shown in Fig. 10 are images of LDH-stearate (Tween 60) crystals at different temperatures as observed with a hot stage microscope. Noteworthy is the fact that the crystals appear superficially intact at temperatures exceeding the second endotherm peak.

DSC melting endotherm and hot stage microscopy of LDH-stearate (Tween 60)
Temperature-scanned XRD
Figures 11–13 show the evolution of the XRD spectra as a function of temperature for LDH-stearate. The abrupt shift in the position of the XRD peaks above ca. 85 °C in Fig. 11 indicates a phase change. This observed shift coincides with the onset of the first melting endotherm observed in the DSC scans (Figs. 9 and 10). The phase change is associated with a decrease in the clay d-spacing from ca. 5.1 nm to about 4.7 nm and an increase in the separation between the alkyl chains from 0.406 nm (2θ = 25.42°) to about 0.425 nm (2θ = 24.6°). In addition, the change in position of the latter peak is associated with some peak broadening suggesting a transition towards disorder. Figure 12 plots the effect of temperature on the corresponding peak intensities for LDH-stearate (SDS). It confirms that the onset temperature for the transition is ca. 85 °C and shows that the new phase disappears at ca. 125 °C. Figure 11 indicates that the material is essentially amorphous at 135 °C except for the LDH-CO3 impurity. This phase slowly disappeared at higher temperatures. Slow cooling to ambient did not recover the highly crystalline LDH-stearate phase. Rather, the material remained amorphous and did not relax back to an ordered state on cooling. Borja and Dutta [17] attributed the disordering of the bimolecular film to the formation of kinks and gauge blocks in the alkyl chains. Additionally, the present DSC results suggest that the chains actually assume a liquid-like state. A measure of recovery is noted when the upper temperature is limited to 100 °C and the material is subsequently allowed to cool down (see Fig. 13). Similar behavior was observed for LDH-laurate. In this case the intermediate phase was observed in the temperature window 65–95 °C.

The effect of temperature on the X-ray diffractogram of LDH-stearate (Tween 60). Scans were taken at 5 °C intervals

The effect of temperature on the intensity of selected X-ray diffraction peaks of LDH-stearate (SDS). Scans were taken at 5 °C intervals

Changes in the X-ray diffractogram on heating LDH-stearate (SDS) to 100 °C and cooling back to ambient. Scans were taken at 5 °C intervals
Conclusion
Molten stearic acid, when dispersed in aqueous media, reacts with LDH-CO3 under atmospheric conditions to form bilayer-intercalated LDH. This implies that, in the presence of water, stearate ions are able to displace the carbonate ions in the layered double hydroxide galleries. The greater stability of bilayer intercalated stearate, compared to LDH-CO3, possibly arises from the stabilizing effect of hydrophobic interactions between the chains [31, 53].
Adding the anionic surfactant SDS positively influenced the intercalation process. It acts as a dispersant for the hydrotalcite particles and keeps unreacted stearate in emulsion thereby also facilitating its removal from the final product. The result is that purer LDH-carboxylate end products are obtained with less mixing and purification effort.
Bilayer intercalated products were previously obtained by exchanging the Cl− in LDH-Cl with, respectively, fatty acids in ethanol and sodium carboxylates in aqueous media [17–19]. The close agreement between the XRD and FTIR analyses of the present products with those previously obtained [17] suggests that the acid intercalated in both the dissociated (R-COO−) and undissociated (R-COOH) forms. Kanoh et al. [18] were unable to intercalate bilayers of decanoic and octanoic acid. This was attributed to the rather high hydrophilicity of the sodium salts used in their method. In contrast, the present procedure employs emulsified neat acids. No problem was encountered in intercalating octanoic acid in bilayer format.
The surfactant-mediated intercalation method is an environmentally friendly option: The SDS solution should be recyclable and no volatile or flammable organic solvents are necessary to free the product from excess stearic acid. However, XRF analysis reveals that the use of SDS also leads to intercalation of some sodium carboxylate in addition to the other carboxylic acid forms. This problem can be obviated by replacing the SDS with a non-ionic surfactant such as Tween 60.
It was found that the intercalation products obtained with either surfactant showed similar thermal behavior. Specifically, two phase transformations are observed at elevated temperatures. At temperatures that are significantly higher than the melting points of the corresponding free acids, the alkyl chains assume a disordered liquid-like state within the clay galleries. In addition, a decrease in the interlayer spacing is observed. This state is reversible to some extent. However, at even higher temperatures the material becomes completely amorphous and behaves like a true melt. Cooling of this melt does not lead to the recovery of the well-ordered crystalline state. These observations have two important implications. Firstly, once fully molten, it appears that the presence of interlayer water may be required for recrystallization. Secondly, exfoliation of stearate-intercalated clay is unlikely to proceed using conventional polymer melt-blending techniques. This is because the LDH-stearate itself melts below typical polymer processing temperatures. Furthermore, LDH crystals are only stable when they form a stacking structure of at least 20 layers [48].
References
Miyata S (1983) Clays Clay Miner 31:305
Kopka H, Beneke K, Lagaly G (1988) J Colloid Interface Sci 123:427
Bellotto M, Rebours B, Clause O, Lynch J (1996) J Phys Chem 100:8527
Miyata S, Kumura T (1973) Chem Lett 843
Chandler D (2005) Nature 437:640
Whitesides GM, Mathias JP, Seto CT (1991) Science 254:1312
O’Hare D (1991) In: Bruce DW, O’Hare D (eds) Inorganic materials. John Wiley and Sons, New York, p 167
Carlino S (1997) Solid State Ionics 98:73
Crepaldi EL, Pavan PC, Valim JB (1999) Chem Commun 155
Reichle WT (1985) J Catal 94:547
Drezdzon MA (1988) Inorg Chem 27:4628
Raki L, Rancourt DG, Detellier C (1995) Chem Mater 7:221
Whilton NT, Vickers PJ, Mann S (1997) J Mater Chem 7:1623
Aisawa S, Hirahara H, Uchiyama H, Takahashi S, Narita E (2002) J Solid State Chem 167:152
Zhang J, Zhang F, Ren L, Evans DG, Duan X (2004) Mater Chem Phys 85:207
Meyn M, Beneke K, Lagaly G (1990) Inorg Chem 29:5201
Borja M, Dutta K (1992) J Phys Chem 96:5434
Kanoh T, Shichi T, Takagi K (1999) Chem Lett 117
Itoh T, Ohta N, Shichi T, Yui T, Takagi K (2003) Langmuir 19:9120
Dimotakis ED, Pinnavaia TJ (1990) Inorg Chem 29:2393
Miyata S, Okada A (1977) Clays Clay Miner 25:14
Sato T, Mikako T, Endo T, Shimada M (1987) J Chem Technol Biotechnol 39:275
Chibwe K, Jones W (1989) J Chem Soc Chem Commun 926
Crepaldi EL, Tronto J, Cardoso LP, Valim JB (2002) Colloids Surf A 211:103
Hansen HCB, Taylor RM (1991) Clay Miner 26:311
Morioka H, Tagaya H, Karasu H, Kadokawa J, Chiba K (1995) J Solid State Chem 117:337
Carlino S, Hudson MJ (1994) J Mater Chem 4:99
Carlino S, Hudson MJ, Husain SW, Knowles JA (1996) Solid State Ionics 84:117
Prevot V, Forano C, Besse JP (1998) Inorg Chem 37:4293
Prevot V, Forano C, Besse JP (1999) J Mater Sci 9:155
Choy J-H, Kwak S-Y, Park J-S, Jeong Y-J, Portier J (1999) J Am Chem Soc 121:1399
Evans DG, Duan X (2006) Chem Commun 485
Khan AI, O’Hare D (2002) J Mater Chem 12:3191
Vaccari A (1998) Catal Today 41:53
Leroux F, Besse J-P (2001) Chem Mater 13:3507
Fisher H (2003) Mater Sci Eng C 23:763
Costa FR, Abdel-Goad M, Wagenknecht U, Heinrich G (2005) Polymer 46:4447
Costa FR, Wagenknecht U, Jehnichen D, Abdel-Goad M, Heinrich G (2006) Polymer 47:1649
Costa FR, Leutetriz A, Wagenknecht U, Jehnichen D, Häuβler L, Heinrich G (2007) Appl Clay Sci
Carlino S, Hudson MJ (1995) J Mater Chem 5:1433
Dèkány I, Haraszti T (1996) Colloid Surf A 123–124:391
Pavan PC, Gomes GA, Valim JB (1998) Micropor Mesopor Mater 21:659
Pavan PC, Crepaldi EL, Valim JB (2000) J Colloid Interface Sci 229:346
You Y, Zhao H, Vance GF (2002) Colloids Surf A: Physicochem Eng Aspects 205:161
You Y, Zhao H, Vance GF (2002) J Mater Chem 12:907
Clearfield A, Kieke M, Kwan J, Colon JL, Wang RC (1991) J Incl Phenom Mol Recogn Chem 11:361
Adachi-Pagano M, Forano C, Besse J-P (2000) Chem Commun 91
He JX, Yamashita S, Jones W, Yamagishi A (2002) Langmuir 18:1580
Rey F, Fornés V, Rojo JM (1992) J Chem Soc Faraday Trans 88:2233
Bera P, Rajamathi M, Hegde MS, Kamath PV (2000) Bull Mater Sci 23:141
Vold RD, Hattiangdi GS (1949) Ind Eng Chem 41:2311
Bracconi P, Andres C, Ndiaye A (2003) Int J Pharm 262:109
Takagi K, Sichi T, Usami H, Sawaki Y (1993) J Am Chem Soc 115:4339
Acknowledgements
Financial support for this research, from the Institutional Research Development Programme (IRDP), the South African Cooperation Fund for Scientific and Technological Developments (NEPAD) and the THRIP program of the Department of Trade and Industry and the National Research Foundation of South Africa, as well as Xyris Technology CC, is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nhlapo, N., Motumi, T., Landman, E. et al. Surfactant-assisted fatty acid intercalation of layered double hydroxides. J Mater Sci 43, 1033–1043 (2008). https://doi.org/10.1007/s10853-007-2251-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-007-2251-0