
Abstract
In this paper, the formation of titania nanotube arrays was investigated in NH4F/H2SO4 electrolyte. Under optimized electrolyte conditions, the titania nanotube arrays with an inner diameter of about 120 nm and a length of about 300 nm was obtained. During the formation process, the variety of current was observed. The current–time curve implied that the evolvement process of titania nanotube arrays include three stages. The stability of titania nanotube arrays at elevated temperatures was studied. The as-prepared titania nanotube arrays is amorphous, crystallized in anatase and rutile as the rising of the temperature. The samples were characterized by ESEM and XRD.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Titania is an important inorganic functional material. It has been intensively investigated due to a variety of its potential applications such as transducer, self-cleaning material, solar cells, catalyst and photoelectricity material [1–5]. Ordinary phases of the titania include amorphism, anatase and rutile. The phase and shape of titania determine its properties and potential applications. For example, the anatase phase of titania is preferred in dye-sensitized solar cells and catalysis, whereas rutile is mostly used in the area of dielectrics and high-temperature oxygen gas sensors [6, 7]. Recently, a new pattern of titania with discrete, tubular and well-ordered nanotube arrays was obtained by anodization of titanium in fluorine-containing electrolyte [8–12]. The electrolyte composition and anodic voltage primarily determine the structure of the oxide layer. Electrolyte without fluoride such as sulfuric acid has been widely used in anodization of titanium. In these electrolytes, a nonporous compact titania film is formed at low potentials, and porous titania film is formed at high potentials due to electrical breakdown of the oxide [13–16]. In fluoride-containing electrolyte, highly ordered nanotube arrays, rather than porous or nonporous structures, is formed at low potentials 10–20 V, as a result of the competition between the formation and the dissolution of titania nanotube arrays [9–12]. Until now, according to the reports, the phase of titania nanotube arrays is amorphous and the inner diameter of nanotube is 20–100 nm. Titania nanotube arrays has shown a variety of functional properties for potential applications such as photoelectrochemical materials [17–20], sensors [21–23], catalyst support [24, 25], dye sensitization [26], controlled wetting materials [27, 28], biocompatibility materials [29, 30] and template material [31, 32]. All of the fabrication studies focus on the surface morphology of the titania nanotube arrays; different morphology leads to different application. In order to extend the potential applications of titania nanotube arrays for more fields, the size of the nanotube must be considered in its fabrication.
In the present work, titania nanotube arrays with an inner diameter of about 120 nm and outer diameter of about 140 nm was obtained by anodization of titanium in NH4F/H2SO4 aqueous solution. The heat stability of as-prepared titania nanotube arrays was studied. The evolvement process of the tube was studied by the variety of current–time and the analysis from literatures.
Experimental
Titanium foils (99.5% pure) were purchased from Northwest Institute For Non-ferrous Metal Research (China). Prior to anodization, the titanium foils were ultrasonically cleaned in acetone and distilled water, respectively, for 5 min, then eroded in 4 wt% HF + 5 mol/l HNO3 for 30 s, followed by ultrasonically cleaned in distilled water for 5 min and dried in air at 40 °C. A two-electrode with a graphite cathode was used to fabricate the nanotubes. 0.0015–1.84 mol l−1 H2SO4 and 0.1–2 wt% NH4F aqueous solutions were used as electrolytes. All electrolytes were prepared from analytical grade chemicals and DI water. The anodizing voltage was kept constant during the entire process with a DC power supply (GOA, China). The whole course of anodization was conducted at room temperature (25 °C) with magnetic agitation. The time-dependent variation in current was monitored using a digital multimeter (DT9202, China), once every 5 s. The sample was taken out and washed immediately after 60 min or longer, then dried at 40 °C in air and annealed at different temperature (300–600 °C) for 4 h in furnace with air to research the effect of anneal temperature on the morphology and crystal transformation of the titania nanotube arrays. The same heat treatment experiment was carried on pure titanium as blank.
Philips XL30 ESEM was employed for the structural and morphological characterization of the sample. The cross-section photograph was obtained by observing mechanically fractured sample. X-ray diffraction measurements were performed on Philips X′ Pert MPD diffractometer with CuKα radiation, the X-ray generator operated at 40 kV and 40 mA. Date sets were collected over the range of 5–90° with a step size of 0.02° and a count rate of 4.0°/min. Phases identification was achieved by comparing the diffraction patterns of Ti and TiO2 with ICDD (JCPDS) standards.
Results and discussion
Figure 1a and b shows the surface structures of the samples obtained with different anodizing voltages, 20 and 30 V, in 0.24 mol l−1 H2SO4 + 0.5 wt% NH4F aqueous solution. At low anodizing voltage, 5 and 10 V, only a compact oxide film formed on the surface of titanium. As the voltage increased to 20 V, the surface structures of the titanium become discrete, hollow, cylindrical, and tubular, as shown in Fig. 1a. This tubular structure has an inner diameter of about 120 nm and wall thickness of 25 nm. However, as voltage increased to 30 V, as shown in Fig. 1b, there is no well-ordered titania nanotube arrays on the titanium substrate. It is clear that the anodizing voltage has important effect on the anodization of titanium. The reason is that the formation of the nanotube layer is essentially the result of equilibrium between electrochemical formation of titania and the chemical dissolution of titania in fluoride-containing solution [9–12, 33]. Under different anodizing voltages, different pH profiles were built on the surface of the titanium [33] and the Ti–O bond of the titania occurs different degree of polarization, which lead to different dissolution of the titania. Therefore the titania nanotube arrays is obtained only under appropriate anodizing voltage.

The effect of voltage on the surface of the anodization titanium (0.24 mol l−1 H2SO4 + 0.5 wt% NH4F, 60 min) (a) 20 V; (b) 30 V
Figure 2a and c shows the surface structures of the samples obtained in different concentrations of H2SO4 solution (0.0015–1.84 mol l−1) with 0.5 wt% NH4F at 20 V for 60 min. It is clear, as shown in Fig. 2, that the titania nanotube arrays formed in 1.75 mol l−1 H2SO4 (Fig. 2b) and 0.24 mol l−1 H2SO4 (Fig. 1a), unformed in 0.0015, 0.018 and 1.84 mol l−1 H2SO4. It is remarkable that the inner diameter of nanotube decreases with H2SO4 concentration from 120 nm in 0.24 mol l−1 H2SO4, to 100 nm in 1.75 mol l−1 H2SO4. The difference in concentrations of H2SO4 lead to significant variations of the surface structures of the titanium, indicating that there is an appropriate concentration of H2SO4 for the formation of titania nanotube arrays. Because there is a primary reaction in the anodization of titanium: TiO2 + 6F- + 4H+ → TiF 2−6 + 2H2O [12]. H2SO4 solutions with different concentration possess different acidity, which lead to different chemical dissolution of titania and influence the equilibrium of between electrochemical formation of titania and chemical dissolution of titania. Therefore, no titania nanotube arrays could be formed in the electrolyte, whose acidity is too high or too low.

The effect of H2SO4 concentration on the surface of the anodization titanium (20 V, 0.5 wt% NH4F, 60 min) (a) 0.018 mol l−1 H2SO4; (b) 1.75 mol l−1 H2SO4; (c) 1.84 mol l−1 H2SO4
Figure 3a and b show the surface structures of the samples obtained in 0.24 mol l−1 H2SO4 aqueous solution with various concentrations of NH4F (0.1–2 wt%) at 20 V for 60 min. There is no titania nanotube arrays formed in 0.1 wt% NH4F and 2.0 wt% NH4F (Fig. 3b). When anodized in 0.5 wt% NH4F (Fig. 1a) and 1.0 wt% NH4F (Fig. 3a), titania nanotube arrays were obtained. The results indicated that the ion concentration of the F− could also influence the morphology of the sample and there must exist an optimal value of [F−]/[H+] fitting for the formation of the titania nanotube arrays. Because the chemical dissolution of titania is determined by the F− concentration and solution acidity.

The effect of NH4F concentration on the surface of the anodization titanium (0.24 mol l−1 H2SO4, 20 V, 60 min) (a) 1.0 wt%; (b) 2.0 wt%
In order to assess the influence of time on the surface of the sample, a set of experiments was performed in 0.24 mol l−1 H2SO4 + 0.5 wt% NH4F aqueous solution under 20 V for 60, 180 and 540 min. The cross-section of samples was examined after the experiments with an ESEM. The same thickness was observed (Fig. 4). The result consistent with earlier work [9], which indicates that there is a competition between electrochemical formation of titania and its chemical dissolution, and this dynamic equilibrium controls the thickness of titania layer.

The cross-section of the titania nanotube arrays (0.24 mol l−1 H2SO4 + 0.5 wt% NH4F, 60 min)
Figure 5 shows the current transient recorded for titanium during holding the samples at 20 V for 20 min, in 0.5 wt% NH4F (Fig. 5a), in 0.24 mol l−1 H2SO4 solutions (Fig. 5b), and in 0.24 mol l−1 H2SO4 + 0.5 wt% NH4F solutions (Fig. 5c). In pure H2SO4 or pure NH4F solution, the current decay is observed that can be ascribed to the classical highfield oxide formation behavior, as through ESEM observation, the sample has compact oxide layer on the surface of titanium. For the 0.24 mol l−1 H2SO4 + 0.5 wt% NH4F solution, the current transients deviate clearly from the pure H2SO4 or pure NH4F curve. The current–time curve was separated clearly into 3 stages. After an initial decay, the current starts rising again and keep steadily. Such curves are typical for the formation process of the titania nanotube arrays [10, 34].

Current transient recorded for titanium during anodization (20 V, 20 min) (a) pure 0.5 wt% NH4F; (b) pure 1.75 mol l−1 H2SO4; (c) 0.5 wt% NH4F + 1.75 mol l−1 H2SO4
On the basis of the observation and the reports of the growth mechanism of the titania nanotube arrays [35–37], the evolvement process of nanotube of the titania nanotube arrays is proposed to build a model, which is shown in Fig. 6. In the first stage with the onset of anodization, a thin barrier layer of titania forms and leads to the current decay (Fig. 6a). Due to the solubility of the titania in F-containing acidic solution, the current starts to increase in the second stage. In this stage pores come into being (Fig. 6b) and grow subsequently. The formation of the pores enhances the electric-field across the remaining barrier layer, results in the remaining barrier layer stars to dissolve with high speed and leads to formation of the chimbs between two pores (Fig. 6c). Subsequently, the pores and the chimbs become deeper and deeper (Fig. 6d). Ultimately, both voids and tubes grow in equilibrium and finally yield a tubular structure (Fig. 6e).

Schematic diagram of the evolution of titania nanotubes in anodization as follows: (a) oxide layer formation; (b) pore formation on the oxide layer; (c) chimbs formation between pores; (d) growth of the pores and the chimbs; (e) fully developed of the titania nanotube arrays
The stability of titania nanotube arrays at elevated temperatures, 300–600 °C, was studied in air. No discernible changes in the dimensions of the tubes were found when the heat treatment was performed at 300 °C for 4 h, a little shrinkage in average inner diameter and wall thickness at 500 °C, obvious breakage at 600 °C as shown in Fig. 7.

SEM micrographs of titania nanotube arrays annealed at 600 °C (0.24 mol l−1 H2SO4 + 0.5 wt% NH4F, 60 min)
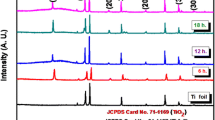
Figure 8 shows the XRD patterns of the sample annealed at different temperatures. The as-prepared titania nanotube arrays were found to be amorphous (Fig. 8e). It is clear that the sample crystallized in the anatase phase at a temperature close to 300 °C (Fig. 8b). The anatase phase was enhanced at 500 °C (Fig. 8c). When the temperature arrived at 600 °C (Fig. 8d), the rutile phase emerges in the X-ray diffraction pattern. In order to testify that the change of the crystallization comes of titania nanotube arrays, the same heat treatment were performed on pure titanium without titania nanotube arrays. There is only Ti peak appears on the pattern at 500 °C. The rutile phase emerges in the X-ray diffraction pattern straightly at 600 °C (Fig. 8a). The results indicate that the crystallization of titania nanotube arrays transformed as the annealed temperature rised. The change of configuration of the titania nanotube arrays at high temperature is the result of crystallization of titania nanotube arrays and oxidation of titanium support.

XRD patterns of the pure titanium with or without titania nanotube arrays annealed at different temperatures (0.24 mol l−1 H2SO4 + 0.5 wt% NH4F, 60 min). (a) Without, 600 °C; (b) with, 300 °C; (c) with, 500 °C; (d) with, 600 °C; (e) with, unannealed
Anatase and rutile are two ordinary phases of the titania; both belong to tetragonal structure. But there are four TiO2 molecules in the crystal cell of the anatase, two in that of rutile. Oxygen octahedrons are joined together via face in anatase, via culmination in rutile. According to the third principle of Pauling, the presence of shared polyhedral edges and especially shared polyhedral faces decreases the stability of a crystal structure [38]. So the crystal phase of titania nanotube arrays could transform from anatase to rutile as the temperature rised.
Conclusions
Well-ordered titania nanotube arrays with an inner diameter of about 120 nm, a length of about 300 nm could be obtained on the surface of pure titanium foils by anodic oxidation in 0.24 mol l−1 H2SO4 + 0.5 wt% NH4F solution at 20 V. The evolvement processes of the titania nanotube arrays include three stages, or five details. The as-prepared titania nanotube arrays is amorphous, crystallize in the anatase phase at a temperature close to 300 °C. The anatase phase is enhanced at 500 °C. When temperature arrives at 600 °C, the rutile phase emerges in the X-ray diffraction pattern.
References
Fujishima A, Honda K (1972) Nature 238:37
Livraghi S, Votta A, Paganini MC, Giamello E (2005) Chem Commun 4:498
Carotta MC, Ferroni M, Gnani D, Guidi V, Merli M, Martinelli G, Casale MC, Notaro M (1999) Sens Actuat B 58:310
Savage NO, Akbar SA, Dutta PK (2001) Sens Actuat B 72:239
Shimizu Y, Kuwano N, Hyodo T, Egashira M (2002) Sens Actuat B 83:195
Hagfeldt A, Gratzel M (2000) Acc Chem Res 33:269
Varghese OK, Gong D, Paulose M, Grimes CA, Dickey EC (2003) J Mater Res 18:155
Zwilling V, Aucouturier M, Darque-Ceretti E (1999) Electrochim Acta 45:921
Gong D, Grimes CA, Varghese OK, Hu W, Singh RS, Chen Zh, Dickey EC (2001) J Mater Res 16:3331
Beranek R, Hildebrand H, Schmuki P (2003) Electrochem Solid-State Lett 6:B12
Mor GK, Shankar K, Paulose M, Varghese OK, Grimes CA (2005) Nano Letters 5:191
Cai Q, Paulose M, Varghese OK, Grimes CA (2005) J Mater Res 20:230
Yang B, Uchida M, Kim HM, Zhang X, Kokubo T (2004) Biomaterials 25:1003
Sul YT, Johansson CB, Jeong Y, Albrektsson T (2001) Med Eng Phys 23:329
Marchenoir JC, Loupet JP, Masson J (1980) Thin Solid Films 66:357
Dyer CK, Leach JSL (1978) J Electrochem Soc 7:1032
Mor GK, Shankar K, Varghese OK, Grimes CA (2004) J Mater Res 19:2989
Beranek R, Tsuchiya H, Sugishima T, Macak JM, Taveira L, Fujimoto S, Kisch H, Schmuki P (2005) Appl Phys Lett 87:243114
Ghicov A, Macak JM, Tsuchiya H, Kunze J, Haeublein V, Frey L, Schmuki P (2006) Nanoletters 6:1080
Xie Q, Yang Sh, Ruan X, Zhao H (2005) Environ Sci Technol 39:3770
Varghese OK, Gong D, Paulose M, Ong KG, Grimes CA (2003) Sens Actuat B 93:338
Mor GK, Carvalho MA, Varghese OK, Pishko MV, Grimes CA (2004) J Mater Res 19:628
Mor GK, Varghese OK, Paulose M, Ong KG, Grimes CA (2006) Thin Solid Films 496:42
Macak JM, Tsuchiya H, Bauer S, Ghicov A, Schmuki P, Barczuk PJ, Nowakowska MZ, Chojak M, Kulesza PJ (2005) Electrochem Commun 7:1417
Chen S, Paulose M, Ruan C, Mor GK, Varghese OK, Kouzoudis D, Grimes CA (2006) J Photochem Photobiol A: Chem 177(2−3):177
Macak JM, Tsuchiya H, Ghicov A, Schmuki P (2005) Electrochem Commun 7:1133
Baluar E, Macak JM, Taveira L, Schmuki P (2005) Electrochem Commun 7:1066
Balaur E, Macak JM, Tsuchiya H, Schmuki P (2005) J Mater Chem 15:4488
Tsuchiya H, Macak JM, Muller L, Kunze J, Muller F, Greil SP, Virtanen S, Schmuki P (2006) J Biomed Mater Res 77A:534
Macak JM, Tsuchiya H, Taveira L, Ghicov A, Schmuki P (2005) J Biomed Mater Res 75A:928
Prida VM, Hernandez-Velez M, Cervera M, Pirota K, Sanz R, Navas D, Asenjo A, Aranda P, Ruiz-Hitzky E, Batallan F, Vazquez M, Hernando B, Menendez A, Bordel N, Pereiro R (2005) J Magn Magn Mater 294:e69
Oh S, Finones RR, Daraio C, Chen L, Jin S (2005) Biomaterials 26:4938
Macak JM, Tsuchiya H, Schmuki P (2005) Angew Chem Int Ed 44:2100
Ghicov A, Tsuchiya H, Macak JM, Schmuki P (2005) Electrochem Commun 7:505
Taveira LV, Macak JM, Tsuchiya H, Dick LFP, Schmuki P (2005) J Electrochem Soc 152:B405
Macak JM, Tsuchiya H, Taveira L, Aldabergerova S, Schmuki P (2005) Angew Chem Int Ed 44:7463
Mor GK, Varghese OK, Paulose M, Mukherjee N, Grimes CA (2003) J Mater Res 18:2588
Rohrer GS (2001) Structure and bonding in crystalline materials. Cambridge University Press, Cambridge, p 441
Acknowledgements
The authors would like to thank National Nature Science Foundation of China (30600149), the science research foundation of ministry of Health & United Fujian Provincial Health and Education Project for Tackling the Key Research, P.R. China (WKJ 2005-2-008) and Fujian Development and Reform Commission of China (No. 2004[477]).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tian, T., Xiao, Xf., Liu, Rf. et al. Study on titania nanotube arrays prepared by titanium anodization in NH4F/H2SO4 solution. J Mater Sci 42, 5539–5543 (2007). https://doi.org/10.1007/s10853-006-1104-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-006-1104-6