
Abstract
The complexation process between racemic flurbiprofen and β-cyclodextrin in solution was investigated by 1D and 2D proton NMR spectroscopy. In the presence of β-cyclodextrin, the aromatic protons of flurbiprofen were the most affected, suggesting a strong involvement of the phenyl groups in the inclusion mechanism. The stoichiometry of the complex was determined by the method of continuous variation, using the chemical induced shifts of both host and guest protons. The association constant, Ka of the obtained complex was calculated and found to be 2483.8 M−1. On the other hand, signals belonging to the protons associated with the carboxyl group are split in the presence of β-cyclodextrin indicating enantiomeric differentiation. Rotating frame NOE spectroscopy, (ROESY), was used to ascertain the solution geometry of the host–guest complex. The result suggested that the flurbiprofen molecule fully penetrates the β-cyclodextrin cavity with the carboxyl group protruding from the primary hydroxyl side and the phenyl group close to the secondary rim.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flurbiprofen, (RS)-2-(2-fluoro-4-biphenyl) propionic acid (FP), is an optically active anti-inflammatory drug (Fig. 1), used to treat the inflammation and pain of arthritis [1]. However, due to its low water solubility, poor absorption characteristics of FP have been reported [2]. Therefore aqueous solubility and wettability of flurbiprofen give rise to difficulties in pharmaceutical formulations for oral or parenteral delivery, which may lead to variable bioavailability. To overcome these drawbacks, increasing the aqueous solubility of FP is an important goal. In order to improve the dissolution rate and oral bioavailability of flurbiprofen, several investigations [3–6], were made using inclusion complexes of FP with naturally occurring α, β and γ-cyclodextrins, or chemically modified ones (Fig. 2).

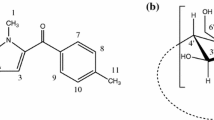
Chemical structure of flurbiprofen. Numerals correspond to proton positions referred to in the NMR study

Numbering of the glucose unit and molecular structures of natural and chemically modified CDs
Cyclodextrins are naturally occurring cyclic oligosaccharides, known for their effect on stability, solubility and bioavailability of various drugs, as well as for the reduction of drugs side-effects [7]. The α-1, 4-linked d-glucopyranose units form a CD ring with hydrophilic hydroxyl groups on their outer surface and a hydrophobic cavity in the center. CDs are capable of forming inclusion complexes with many drugs by taking up a whole drug molecule, or some part of it, into the cavity. Such molecular encapsulation will affect many of the physicochemical properties of the drugs, such as their aqueous solubility and rate of dissolution. During the drug-CD complex formation, no covalent bonds are formed and the complex is readily dissociated and the free drug molecules are in rapid equilibrium with the drug molecules bound within the CD cavity. Complexes between CDs and flurbiprofen were studied by a variety of techniques including X-ray diffraction [8–10], isothermal titration calorimetry and solid state 13C NMR spectroscopy [3, 4], solubility analysis and affinity capillary electrophoresis [5, 6]. Of these techniques, only X-ray diffraction and NMR can yield information about the inclusion complex on an atomic level. X-ray diffraction provides a static view of the complex and depends on the availability of a good-quality single crystal. Because NMR measurements can be performed in solution, this method offers the advantage of providing insights into dynamic processes as well as structural information at a molecular level. Thus 1H NMR titration is one of the most used methods to quantitatively investigate the formation of CD complexes [11, 12]. Complexation of CD with guest molecules causes changes in the chemical shifts of the protons belonging to the cyclodextrin and/or guest molecule. The observed chemical shift changes in 1H NMR titration can provide the conformation of formed supramolecular complex and independent signals for the evaluation of association constant, Ka.
The aim of this work is to gain further insight into the mode of inclusion and stability of the flurbiprofen./β-cyclodextrin in aqueous solution, using 1D and 2D NMR techniques. These techniques are particularly sensitive to small changes in the electronic environment of a proton that occur upon intimate contact or short-range association in the complex. Firstly the stoichiometry of the complex was investigated using the current Job plot method. The type of inclusion process of FP in β-CD was confirmed by 2D ROESY spectroscopy. The association constant of the complex was determined using 1H NMR titration method in solution, followed by a non-linear fitting procedure.
Experimental
Materials
Flurbiprofen and β-cyclodextrin (water content 8 mol/mol) were purchased from Sigma-Aldrich Chemie GmbH and used without further purification. The β-CD water content was taken into account in the calculation of solute concentration. The deuterium oxide (99.7 %D) was obtained from Heavy Water Plant Romag-Prod, Romania.
Sample preparation
In order to study the complexation process between FP and β-CD, two stock solutions in D2O, of 5 mM were prepared. The FP stock solution was prepared containing 5 μL/mL of NaOD 40 % to aid with the dissolution of the drug. Based on these equimolar solutions, a series of samples containing β-CD and FP were prepared. This was accomplished by mixing the two solutions to constant volume at varying proportions so that a complete range (0 < r < 1) of the mole fraction r = [X]/([G] + [H]) was sampled. X = H or G and [H] and [G] are the concentrations of the host (β-CD) and guest (FP), respectively. Thus the total concentration [H]t + [G]t = 5 mM was kept constant for each sample. This set of samples was used both for determination of stoichiometry and the evaluation of the association constant, Ka.
1H NMR measurements
All 1H NMR measurements were carried out on a Bruker AVANCE III spectrometer operating at 500.13 MHz and equipped with a broadband observe probe. The chemical shifts were referenced to TMS. In all experiments the temperature was maintained at 298 K and standard 5 mm NMR tubes were used. For each 1H NMR experiment, between 32 and 256 transients were collected into 65 K data points over a 4,000 Hz spectral window, using a 2 s relaxation delay. The 2D ROESY spectra were acquired in the phase sensitive mode and residual water suppression, using Bruker standard parameters (pulse program roesyphpr). Each spectrum consisted of a matrix of 2 K/4 K data points covering a spectral width of 4,000 Hz. The spectra were obtained with a spin-lock mixing time of 500 ms, relaxation delay 3 s and 8 scans were recorded.
Results and discussion
Determination of the stoichiometry
NMR is a technique, which provides the most evidence for the inclusion of a guest into the hydrophobic β-CD cavity in solution. Inclusion of FP in β-CD cavity is evidenced by the change in chemical shifts of some of the guest and host protons, in comparison with the chemical shifts of the same protons in the free components. Partial 1H NMR spectra of pure components and flurbiprofen/β-CD mixtures are shown in Figs. 3 and 4. The absence of new peaks that could be assigned to the complex, suggested that complexation is a dynamic process, the included FP being in a fast exchange between the free and bound states.

Partial 1H spectra (only the aromatic protons) of FP. a 5 mM FP and b 4 mM FP and 1 mM β-CD

Partial 1H NMR spectra (without H1′ proton) of β-CD. a 5 mM β-CD and b 3 mM β-CD and 2 mM FP
When comparing spectrum 3(a) with 3(b) it is possible to observe a subtle signal broadening and chemical shifts variation for some aromatic protons of flurbiprofen, due to the presence of β-CD. As expected, the H3′ and H5′ protons located inside the cavity are appreciably shifted (see Fig. 4), evidencing the existence of an interaction between the flurbiprofen molecule and the interior of the β-CD cavity. The observed broadening may be due to the restricted motion of a flurbiprofen molecule inside a β-CD cavity and by the complexation association–dissociation process. The 1H chemical shifts variation of some representative β-CD and FP protons as a function of their concentration is presented in Fig. 5. It can be seen that in the presence of β-CD, almost all guest protons resonances were affected.

Chemical shifts variation of some representative β-CD and FP protons, as a function of their concentration
The aforementioned 1H NMR observations allow us to use the continuous variation method to determine the stoichiometry of the formed complex. In our case, the continuous variation method is based on the induced chemical shift variation, Δδ, which is directly related to the concentration of the complex. Δδ is defined as the difference in chemical shifts in the absence and in the presence of the other reactant. Thus if a physical quantity, containing Δδ, is plotted as a function of the mol fraction of the host or guest, r, (Job’s plot), its maximum value will occur at r1 = m/(m + n) or r2 = n/(m + n), where m and n are, respectively, the molar ratios of β-CD and drug in the (drug)n:(β-CD)m complex. Under fast exchange conditions, for a signal belonging to β-CD, for example, the calculated quantity Δδ·[β-CD] is proportional to the complex concentration and, can be plotted against r1, [13]. The continuous variation method was applied for protons belonging both to guest and host molecules and yielded identical results. For the sake of concision, only several protons (the most markedly affected) have been selected and reported in Fig. 6.

Job’s plots corresponding to the induced chemical shift variation of some a β-CD and b FP protons, for the FP:β-CD system
In all cases, Job’s plots show a maximum at 0.5, indicating the existence of a complex with 1:1 stoichiometry.
ROESY experiments
While the 1D NMR provides unambiguous evidence on the formation of a complex, ROESY experiments provide information on the dynamics and the averaged relative inter- and intramolecular proton distances. In the present study, to gain further information on the inclusion complexation mode and additional insights into the dynamic structure, a 2D ROESY 1H NMR spectrum was acquired. Due to the rapid dynamics of the complexation process, the ROESY effects were only qualitatively used and no conclusions on intermolecular distances were extracted. An expansion of the ROESY spectrum of the FP:β-CD complex is reported in Fig. 7.

Expanded region of the ROESY spectrum of FP:β-CD complex. [FP] = [β-CD] = 2.5 mM
The 2D NMR spectrum shows several intermolecular cross-peaks between H3′, H5′ and H6′ protons of β-CD and protons of both aromatic rings of FP, demonstrating the inclusion of these groups in the β-CD cavity. In fact no strong correlations were found between fluorophenyl ring and H3′ protons, suggesting that the fluorophenyl ring is only partially included. The correlation of the phenyl ring protons with H3′ and H5′ was more intense, confirming that this part of the guest molecule is inserted within β-CD cavity through the narrower end of the cavity. Also we found intermolecular cross-peaks only with the inner β-CD protons, which establish intracavity binding without evidence for outside contributions. Based on these findings, the geometrical structure of the FP:β-CD inclusion complex, having 1:1 stoichiometry, can be schematically presented as shown in Fig. 8.

Scheme of proposed FP:β-CD complex inclusion geometry
The proposed structure differs very slightly from the crystal structure of β-CD complex with racemic FP, as determined by Uekama et al. [8], in a X-ray diffraction study. They found that two β-CD molecules were associated through intermolecular hydrogen bonds with their secondary hydroxyl groups, to form a head-to-head dimer. R- and S- isomers of FP molecules are separately included in the two β-CD cavities of the dimer, with the carboxyl group protruding from the primary hydroxyl side. Such 2:2 complex can exist in solid state, but is less probable to be formed in aqueous solution, where the complexation is a dynamic process with the FP molecule being in a state of fast exchange between free and included forms. The 1:1 stoichiometry of the FP:β-CD complex in aqueous solution was also confirmed by the classical phase-solubility procedure [6].
Evaluation of the association constant
In order to determine the extent of the intermolecular binding between FP and β-CD, the association constant has been evaluated. The association constant, Ka, for the 1:1 complex was evaluated by a nonlinear least-squares curve fitting regression analysis of the observed chemical shift changes of the guest and β-CD NMR lines, as a function of concentration according to the following equation [14]
where i counts the sample number and j the studied proton. If the studied proton belongs to the guest or host molecule, then X = G or H, respectively. Δδ (j)c represents the chemical shift difference (for a given proton, j) between the free component and the pure inclusion complex. Equation (1) involves no approximations and correlates the total concentrations of the guest and host molecules with the observed difference in the chemical shift:
We developed a computer program [15], based on an iterative procedure following specific algorithms in order to fit the experimental values of Δδ(i,j) to the appropriate equation. Each iteration step sets up a quadratic function to determine the direction of the search and calculates the error function, E, defined as:
The fitting procedure reaches convergence when the difference between two consecutive E values is less then 10−6. The treatment of the entire set of protons studied produces a single Ka value for the entire process and a set of calculated \( \Updelta \delta_{c}^{\left( j \right)} \) values. The program is quite flexible as both the host and the guest can be observed for spectroscopic perturbations, allowing up to 15 protons to be used in the fitting process. In our case, we applied Eq. (1) for a set of protons consisting in H3′ and H5′ of β-CD and H5; H6,10; H7,9 and H8 of fluorbiprofen. The obtained association constant, using the above described procedure is Ka = 2483.8 M−1 with the error function E = 1.87·10−3 and a correlation factor R = 0.9982. The complete set of chemical shifts in the free state and in the pure complex is reported in Table 1.
It is worth mentioning that a large diversity can be found in the reported values of the association constant, Ka, as is presented in Table 2. In principle, this situation can in part be attributed to the fact that the binding process has been examined by many different experimental methods and at different pH values. Another cause of inconsistency can be the approximations used to determine Ka in methods based on linear least-squares regression analysis.
It is well known [11] that different linearization methods are based on some approximations, which are not always either possible or mathematically correct. Nowadays, the necessity of a carefully nonlinear regression analysis of the experimental data is well established [12], whatever technique may be used, when accurate and meaningful association constants are required.
Enantiomeric differentiation
β-CD can form diastereoisomers by including optically active guests within the cavity of its macrocycle. This effect was shown for a β-CD complex with racemic flurbiprofen in solid state [10], where the R-isomer was included in a similar manner to that of S-isomer in the S-flurbiprofen complex, but the hydrogen-bonding contact involving the carboxyl group was different. The β-CD molecule discriminates between R- and S-flurbiprofen through such differences in hydrogen bonds, and includes each isomer independently. It should be noted that the biphenyl moiety of both R- and S-flurbiprofen shows the R-configuration in these β-CD complexes. This indicates that in solid state, β-CD includes the biphenyl moiety with the R-configuration more favorably than that with S-configuration. In this study, a similar enantiomeric differentiation effect was observed in solution. When the complex is formed, the CH3 protons signal split, as can be seen in Fig. 9. The same splitting effect, (Fig. 10), is observed for the H2 proton, indicating in both cases, enantiomeric differentiation. This enantio-discrimination effect, manifested by the splitting of some NMR signals is a consequence of the coexistence in liquid phase of two diastereomeric host–guest complexes.

1H NMR spectra of flurbiprofen CH3 protons: a pure FP, b FP:β-CD with [FP]/[β-CD] = 3.5/1.5, c FP:β-CD with [FP]/[β-CD] = 2/3

Partial 1H NMR spectra (only the H2 proton), for: a pure FP, b FP:β-CD with [FP]/[β-CD] = 3.5/1.5, c FP:β-CD with [FP]/[β-CD] = 3/2
For the FP:β-CD complex, and in respect to the spectrum, the left and right signals will be labeled (CH3L, H2L) and (CH3R, H2R) and they correspond to the left and right enantiomers, respectively.
The observed enantiomeric differentiations on these protons allowed us to estimate the apparent association constant, KL and KR, for both enantiomers. Because the H5′ signal of β-CD overlap for some FP concentrations the H2 signal of FP, we used only the CH3 signal for obtaining the KL and KR. We used the term apparent for these association constants, due to the fact that the carboxyl group protrudes from the primary hydroxyl side of β-CD and CH3 and H2 protons cannot interact with H3′, H5′ protons of β-CD. They can only stabilize the host–guest complexes through van der Waals contacts with the CH2OH groups of the β-CD. The apparent association constants were determined employing the above mentioned non-linear procedure, in agreement with Eq. (1). The obtained results are KL = 712 M−1 and KR = 146 M−1. This result indicates that complexes formed by left enantiomers are more stable than complexes formed by right enantiomers. We also noticed that FP protons located inside the β-CD cavity are not split, meaning that both isomers are included in a similar manner, in agreement with the solid state structure [10] of the FP:β-CD complex.
Conclusions
The complexation between racemic FP and β–cyclodextrin has been investigated in solution by 1D and 2D 1H NMR spectroscopy. The 1:1 stoichiometry and association constant for the inclusion complex was deduced by NMR titration. The NMR results showed that the biphenyl moiety of the flurbiprofen molecule was deeply penetrated into the β-CD cavity with the carboxyl group outside the primary hydroxyl rim of β-CD. The geometrical structure of the FP:β-CD complex in solution resemble with the crystal structure of the racemic FP complex with β-CD. It was found that complexation of flurbiprofen with β-CD produced a remarkable splitting of the H1 and H2 protons of FP, due to enantio-discrimination. The apparent association constants for the left and right enantiomers were determined.
References
Fukuhara, A., Imai, T., Inoue, K., Otagiri, M.: Effect of oral multiple-dose administration of anti-inflammatory flurbiprofen chimera drug on gastric lesion, other toxicities and disposition kinetics. Biol. Pharm. Bull. 18, 140–147 (1995)
Otagiri, M., Imai, T., Matsuo, N., Uekama, K.: Improvements to some pharmaceutical properties of flurbiprofen by β-and γ-cyclodextrin complexations. Acta Pharm. Suec. 20, 1–10 (1983)
Imai, T., Otagiri, M., Saito, H., Uekama, K.: Inclusion mode of flurbiprofen with β-cyclodextrin and improvements of some pharmaceutical properties of flurbiprofen by complexation. Chem. Pharm. Bull. 36, 354–359 (1988)
Waters, L.J., Bedford, S., Parkes, G.M.B., Mitchell, J.C.: Influence of lipophilicity on drug-cyclodextrin interactions: a calorimetric study. Thermochim. Acta 511, 102–106 (2010)
Imai, T., Irie, T., Otagiri, M., Uekama, K.: Comparative study on inclusion complexation of anti-inflammatory drug flurbiprofen with β-cyclodextrin and methylated-cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2, 597–604 (1984)
Cirri, M., Maestrelli, F., Orlandini, S., Furlanetto, S., Pinzauti, S., Mura, P.: Determination of stability constants values of flurbiprofen-cyclodextrin complexes using different techniques. J. Pharm. Biomed. Anal. 37, 995–1002 (2005)
Loftsson, T., Brewester, M.E.: Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 85, 1017–1025 (1996)
Uekama, K., Hirayama, F., Imai, T., Otagiri, M., Harata, K.: Crystal and molecular structure of 2:2 (±) flurbiprofen-β-cyclodextrin complex. Chem. Pharm. Bull. 31, 3363–3365 (1983)
Uekama, K., Imai, T., Hirayama, F., Otagiri, M., Harata, K.: X-ray crystallographic determination of the absolute configuration of (+) flurbiprofen utilizing β-cyclodextrin complexation. Chem. Pharm. Bull. 32, 1662–1664 (1984)
Harata, K., Uekama, K., Otagiri, M., Hirayama, F.: Crystal structures of cyclodextrin complexes with chiral molecules. J. Incl. Phenom. Macrocycl. Chem. 2, 583–594 (1984)
Fielding, L.: Determination of association constants (Ka) from solution NMR data. Tetrahedron 56, 6151–6170 (2000)
Hirose, K.: A practical guide for the determination of binding constants. J. Incl. Phenom. Macrocycl. Chem. 39, 193–209 (2001)
Djedaïne, F., Lin, S.Z., Perly, B., Wouessidjewe, D.: High-field nuclear magnetic resonance techniques for the investigation of a β-cyclodextrin: indomethacin inclusion complex. J. Pharm. Sci. 79, 643–646 (1990)
Bogdan, M., Caira, M.R., Bogdan, D., Morari, C., Farcas, S.I.: Evidence of a bimodal binding between Diclofenac-Na and β-CD in solution. J. Incl. Phenom. Macrocycl. Chem. 49, 225–229 (2004)
Floare, C., Balibanu, F., Bogdan, M.: Consteq–A program for the calculation of the equilibrium constants, using spectroscopic data. Studia UBB, Physica, Special Issue, L4a, 451–454 (2005)
Acknowledgments
This work was financially supported by UEFISCDI Romania, Project PCE-2011-3-0032.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pirnau, A., Floare, C.G. & Bogdan, M. The complexation of flurbiprofen with β-cyclodextrin: a NMR study in aqueous solution. J Incl Phenom Macrocycl Chem 78, 113–120 (2014). https://doi.org/10.1007/s10847-012-0277-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-012-0277-7