
Abstract
The enantiomer-specific characterization of ofloxacin–cyclodextrin complexes was carried out by a set of complementary analytical techniques. The apparent stability constants of the ofloxacin enantiomers with 20 different cyclodextrins at two different pH values were determined to achieve good resolution capillary electrophoresis enantioseparation either to establish enantioselective drug analysis assay, or to interpret and design improved host–guest interactions at the molecular level. The cyclodextrins studied differed in the nature of substituents, degree of substitution (DS), charge and purity, allowing a systematic test of these properties on the complexation. The seven-membered beta-cyclodextrin and its derivatives were found to be the most suitable hosts. Highest stability and best enantioseparation were observed for the carboxymethylated-beta-cyclodextrin (DS ~ 3.5). The effect of substitution pattern (SP) was investigated by molecular modeling, verifying that SP greatly affects the complex stability. Induced circular dichroism was observed and found especially significant on carboxymethylated-beta-cyclodextrin. The complex stoichiometry and the geometry of the inclusion complexes were determined by 1H NMR spectroscopy, including 2D ROESY techniques. Irrespective of the kind of cyclodextrin, the complexation ratio was found to be 1:1. The alfa-cyclodextrin cavity can accommodate the oxazine ring only, whereas the whole tricyclic moiety can enter the beta- and gamma-cyclodextrin cavities. These equilibrium and structural information offer molecular basis for improved drug formulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
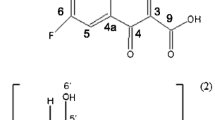
Ofloxacin (OFL), chemically known as (±)-9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylic acid (Fig. 1), belongs to a fluoroquinolone class of chemotherapeutic agents with a broad spectrum of activity against gram-positive and gram-negative bacteria in vivo and in vitro [1–3]. OFL is a racemic compound; the antibacterial activities of the isomers are different. The effect of the S-(−) isomer is 128 times exceeding that of the R-(+)-isomer [4, 5]. The eutomer S-(−) isomer, levofloxacin (LEV) is also available on the drug market as a single enantiomer drug. Separation of the two OFL enantiomers is therefore substantial as shown by several studies. The most common separation method is a chiral ligand-exchange chromatography on a reversed phase HPLC column [6–9]. The other widely used technique is the chiral capillary electrophoresis (CE) using various cyclodextrins (CD) as chiral selectors [10–13].

The structure of native β-CD (left) and the numbered formula of the anionic OFL (right)
CDs, the cyclic oligosaccharides are composed of 6, 7 or 8, α-(1 → 4)-linked glucopyranose units in the respective α-, β- or γ-CDs, depicted often as a truncated cone (Fig. 1). The H3, H5 and glycosidic oxygens are located inside the moderately hydrophobic cavity, while H1, H2, H4, and H6 are in the hydrophilic outer surface of the CD. As a consequence of these features CDs can encapsulate molecules inside their cavity through non-covalent interactions (electrostatic, van der Waals, hydrophobic, hydrogen bond) to form inclusion complex of host–guest type [14–16]. Nowadays not only drugs in native CD complex forms are available in the drug market but drugs in modified, ionic CD complexes such as voricinazole in sulfobutyl-ether-β-CD [17, 18]. CD complexation of pharmaceuticals can result in improved properties of the guest, such as solubility, stability, masking of undesirable properties, protection against oxidation, light-induced reactions and loss by evaporation [19, 20]. Hence the optimized OFL-CD complexes are promising candidates to develop new OFL formulations of improved quality. Beside the enantioseparation, solubility and light-induced decomposition of OFL can also be beneficially modified by CDs. Despite the above advantages of CDs, no literature data appeared on the systematic characterization of OFL-CD using a large number of CDs to obtain equilibrium and structural information, important in the drug formulation process. So far, only the β- and hydroxypropyl-β-CD-OFL complexes were characterized in detail. Koester et al. [21] determined that OFL encapsulation with β-CD increased the solubility 2.6 times but the rate of the photodegradation was not reduced. They determined the apparent 1:1 stability constant by a solubility method which was found to be K = 152 at pH 8.3. Their 1H NMR and molecular modeling study showed that the N-methyl-piperazine ring is located in the inner cavity of the CD. Li et al. [22] prepared and characterized the β- and hydroxypropyl-β-CD-OFL complexes. They determined the apparent 1:1 stability constant at 3 different pHs by fluorescence method, finding K = 1640 and K = 500 for β-, and hydroxypropyl-β-CD at pH 7.53, respectively. Their 2D NOESY NMR studies showed that the tricyclic part of the molecule is in the CD-cavity, while the piperazine moiety is located outside. It can be seen that some data in the literature are contradictory ([21] vs. [22]).
Our aim was to characterize the OFL-CDs complexes by various techniques at two pHs, where the respective average charge of OFL is +1 and −1 [23], and thorough characterization of the complexes by CE, circular dichroism, molecular modeling, 1D and 2D NMR techniques. Here we report the equilibrium and structural parameters of the inclusion complexes formed between 20 CDs and two OFL enantiomers in two charged forms, and the resulting optimized separation of R- and S-OFLs.
Materials and methods
Materials
All native CDs and their derivatives (Table 1) were the products of Cyclolab R&D Ltd. (Budapest, Hungary). Racemic OFL was purchased from Smruthi Organics Ltd. (Solapur, India), LEV (S-OFL) was obtained from Sigma-Aldrich Co. (St. Louis, USA). H3PO4, KH2PO4 Na3PO4, NaOH and HCl were used for buffer preparation using chemicals of analytical grade from commercial suppliers. DMSO, the electro-osmotic flow (EOF) marker in the CE experiment was purchased from Reanal (Budapest, Hungary). D2O was obtained from Sigma-Aldrich. All solutions were prepared from bidistilled Millipore water (specific conductivity: 1.1 μScm−1).
CE experiments
CE experiments were performed on a Hewlett Packard 3DCE instrument equipped with a photodiode array detector. An untreated fused-silica capillary from Agilent was used. Conditioning of new capillaries was conducted by flushing with 1 M NaOH for 30 min followed by 0.1 M NaOH and buffer for 60 min each. The capillary cassette temperature was set to 25 °C and the voltage to +20 kV. UV detection was performed at 200, 210, 230 and 254 nm and samples were run in triplicate. Between the measurements capillaries were flushed with water (1 min), 0.1 M NaOH (0.5 min), water (1 min) and BGE (3 min). The running buffers were PO4 3−/HPO4 2− at pH 11 and 0.05 M phosphoric acid/H2PO4 − at pH 3.5. The ionic strength was constant 0.15 M in both cases. The stock OFL solution (1 mM) contained 0.001 % (v/v) DMSO as EOF marker. Samples were injected hydrodynamically at a pressure of 50 mbar for 3 s.
NMR experiments
All NMR measurements were carried out on a Varian DDR spectrometer (599.9 MHz for 1H) with a 5 mm inverse-detection gradient probehead. Standard pulse sequences and processing routines available in VnmrJ 2.2C/Chempack 4.0 were used. All NMR experiments were carried out in D2O (15 mM phosphate buffer). The chemical shifts were referenced to internal methanol (δ = 3.300 ppm). The average extent of penetration and the direction of inclusion were investigated by two dimensional phase-sensitive rotating frame nuclear Overhauser effect spectroscopy (ROESY). In ROESY experiments the samples contained 1 mM CDs and 2 mM OFL at pH* 3.5. Different mixing times (300 ms, 500 ms) were applied in these experiments.
Circular dichroism measurements
The spectra were recorded on a Jasco J720 Spectropolarimeter using different cylindrical quartz cells. The spectra were accumulated three times with a bandwidth of 1 nm and a scanning step of 0.2 nm at a scan speed of 50 nm/min. In this experiment the samples contained 1 mM OFL and 2 mM various CDs.
Molecular modeling
Modeling of α-CD-OFL, β-CD-OFL, HPβCD-OFL DS4 with 3 different substitution patterns and HPβCD-OFL DS6 complexes were performed using MMFF94 force field implemented in TINKER program. The initial structures of the CDs were based on structural data found in the literature [24] and on structures 2ZYM and 3CGT in the Brookhaven Protein Database [25, 26]. OFL was placed into the cavity at its wider rim in two orientations: either with the carboxyl group or the N-methyl-piperazine group inside. Each structure was subject to energy minimization with 0.001 RMS gradient criteria, simulating aqueous environment with setting ε = 78.3. Molecular dynamic calculations were performed using the Velocity-Verlet algorithm in NVT ensemble with 30 Å sized cubic box periodic boundary conditions, T = 298 K (10000 steps, with 1.0 fs step increment and 0.1 ps snapshot interval).
The resulting 100 structures/guest orientation/charge state/CD were re-optimized and according to the energy values of the optimized structures, the lowest energy ones were taken into account for the interaction energies. The calculated interaction energy was the difference between the energy of the complex and the sum of the energy of the host and the guest.
Results and discussion
CE experiments: stability constants and enantioseparation study
The binding constants between the analyte and the chiral selector are of fundamental interest to understand the inclusion behavior (information on the analyte–ligand affinity and understanding the molecular interactions) [27, 28]. The crucial requirement of CE in binding analysis is that at least one of the interacting species has to carry a charge. Assuming 1:1 averaged stoichiometry for the complex between the host CD and the guest, the effective mobility depends on the CD concentration according to Eq. (1) [29].
where μ eff is the effective mobility of the guest at the actual CD concentration, μ free and μ cplx are the effective mobilities of the free and complexed ligands, respectively, while K is the apparent averaged complex stability constant.
Apparent complex stability constants were determined according to the x-reciprocal method by plotting the data in the form of \( \mu_{\text{eff}}^{\text{i}} - \mu_{\text{free}} / [ {\text{CD ]}} \) versus \( \mu_{\text{eff}}^{\text{i}} - \mu_{\text{free}} \), yielding −K as slope. Beside the x-reciprocal method several other linearization functions are published in the literature [30, 31]. However the x-reciprocal one is a simple, fast and robust method which is ideal for fast screening to determine a large number of binding constants. Due to Wren’s theory, the optimal CD concentration for the enantioseparation can be calculated as:
where K R and K S are the stability constants of the inclusion complexes of the R and S enantiomers, respectively [32].
For the determination of the binding constants, various concentrations of CDs (ranging from 3 to 50 mM) were added to the running buffers. The estimated stability constants and enantioresolution values (calculated at the optimal concentrations according to Wren’s theory) are compiled in Table 2. Resolution of the enantioseparation was calculated by the following equation:
where t 1 and t 2 are the migration times of the enantiomers, and \( w_{1} \) and \( w_{2} \) are the extrapolated peak widths at the baseline.
Table 2 shows that the most stable complex with the native CDs is formed with β-CD, indicating that it has the most suitable cavity size for OFL. The decreasing trend in stability values follows the β-, α-, γ-CD order for all derivatives. For native CDs higher stability constants were measured at acidic pH. This observation shows that the –COOH form of OFL is more appropriate for the complexation than the anionic carboxylate form. The methylation of native CDs decreased the stability constants in our investigations; however the hydroxypropyl substitution (DS3) slightly increased the stability. This can be explained by assuming more secondary bonds between the host and guest molecules. Higher degree of substitution (DS6.3) decreased the stability as the substituents inhibited the inclusion process. Among the neutral CDs, only the HPα- and HPβCD derivatives were able to resolve the OFL enantiomers. Baseline enantioseparation could be achieved with each HPβCD. The highest enantioresolution, however, could be observed for HPβCD DS6.3. The most stable complexes and the best enantioresolution were detected with negatively charged CDs especially CMβCD at pH 3.5. In this inclusion complex additional ionic interaction can develop between the host and guest molecules. The relatively high stability of the MAβCD OFL complex at pH 8 can also be interpreted in terms of ionic interaction but no enantioseparation could be achieved with the positively charged host. It is noteworthy that R-OFL showed stronger binding in each OFL-CD system.
Based on these observations the enantioseparation was developed with CMβCD to achieve high resolution in 10 min, optimizing the buffer pH, concentration, CMβCD concentration, applied voltage, and the injection parameters. Figure 2 shows the electropherogram of the optimized enantioseparation. This enantioseparation could serve as a starting point in the development of a new enantioselective method for the determination of OFL enantiomers in pharmaceutical formulations.

Optimized enantioseparation of OFL enantiomers with CMβCD (electrolyte 20 mM phosphate buffer with 3 mM CMβCD pH = 4.4, applied voltage 15 kV; injection: 50 mbar for 4 s; total length of the capillary 30 cm (21.5 cm effective length), the OFL concentration was 50 μg/ml.)
Molecular modeling study
By molecular modeling the accordance between the computer calculation and the experimental data and the complexation behavior of CD with different substitution patterns was investigated.
The binding energy between different neutral CDs and two different protonated forms of OFL (H2OFL+, OFL−) was investigated by molecular modeling. The values of the energies are summarized in Table 3.
For α-CD and β-CD good correlation was found between the experimental observations and the calculated energy values. Two different degrees of substitution were investigated in the case of HPβCD. The energy values were in agreement with the observations in the CE section: an increased degree of substitution decreased the stability of the inclusion complex. This result points out that molecular modeling can predict even subtle binding affinity differences between host and guest molecules [33]. Thus prediction can reduce the costs of encapsulation development. The hydroxyalkylation of native CD results in mixture of positional isomers, therefore molecular interaction with hosts possessing well-defined substitution pattern may be difficult to study experimentally. In the in silico part of our work three different substitution patterns of HPβCD DS4 were also investigated. Four hydroxypropyl groups were attached to β-CD primary hydroxyl groups in various positions as shown by earlier works [34, 35]. The structure of these three HPβCDs with different substitution patterns can be seen in Fig. 3. These CDs are hydroxyalkylated only at the primary side, which compounds are hardly available by synthesis due to concomitant hydroxyalkylation on the secondary hydroxyl groups. However, these hypothetical guests are suitable for in silico investigation on the role of substitution pattern on guest complexation.

The structure of the three HPβCDs with different substitution patterns (HPβCD DS4a, HPβCD DS4b and HPβCD DS4c, respectively)
The interaction energy values of these complexes show that the substitution pattern is an important factor influencing complex stability as earlier studies also proved experimentally [36, 37]. The complex formation among the HPβCDs is most favorable energetically in the case of HPβCD DS4c (see Table 3). In this CD the substituents are in each other’s closest vicinity so the molecule is not overcrowded and OFL can slip into the CD cavity.
Circular dichroism study
It is known that native CDs are inactive in the UV–VIS absorption region. Nevertheless, an achiral chromophore guest may exhibit an induced circular dichroism (ICD) upon complex formation. The magnitude and direction of ICD signs depend on the complex stability, the dynamics within the complex, the extent of the chromophore inclusion in the CD cavity and the orientation of its electronic transition moment relative to the n-fold rotation axis of CD [38, 39].
In this investigation α-, β-, γ-, HPβ and CMβCD complexes were examined. In our study significant ICD signal could be observed for CMβCD only as can be seen in Fig. 4. This fact is in accordance with the low complex stability of γ- and α-CDs as well as the magnitude of inclusion of α-CD (see NMR section). The main reason of the very low ICD signal intensity with β- and HPβCDs relies in the unfavorable dynamics within the complex. Matsuura et al. observed [40] that rigid, tricyclic analogues of benzophenone, like fluorenone and anthrone, which are similar to OFL gave a very weak ICD while benzophenone can attain a twisted (optically active) conformation within the β-CD cavity. A larger positive ICD can be observed between the OFL and CMβCD due to the enhanced complex stability and the ionic interaction. This positive ICD sign indicates that the aromatic ring system is situated in the CD cavity in axial orientation, in agreement with the Harata rules [41].

The induced circular dichroism signals of OFL and its CD complexes
NMR studies
Job plot titration
The continuous variation method was adopted to verify the stoichiometry of the complexes. The 1H-NMR chemical shifts were measured at different [OFL]/[CD] ratios while the total [OFL] + [CD] was kept constant. The calculated quantities (Δδ [OFL] or [CD]) were plotted as a function of molar ratio. The resulting plots showed a maximum at 0.5 indicating 1:1 binding stoichiometry (invariantly of the actual CD derivative). These results confirm the previously reported findings of Li et al. by fluorescence method [22]. Representative Job plot curves of the two OFL enantiomers and the CMβCD are shown in Fig. 5.

Representative job plot curves of the two OFL enantiomers with CMβCD
1H NMR study
The inclusion complex formation can be proved from the 1H NMR chemical shift changes. Figure 6 illustrates the aromatic peak changes of OFL upon complexation with CMβCD. It can be seen that at higher molar ratios of CMβCD the aromatic protons of the two enantiomers are separated. This method can be suitable to distinguish the OFL enantiomers. The 1H chemical shifts of OFL in the absence and presence of 8 different CDs are indicated in Table 4.

Aromatic region of OFL 1H NMR spectrum at pH* 3.5 (bottom) and the same region in the presence of 10 mM CMβCD (top)
Table 4 shows that complexation-induced chemical shift changes occur most with β-CDs due to the high complex stability. H5, H2 and H13 protons appear at higher chemical shifts as opposed to H2′, H6′, H3′, H5′ and H15 piperazine protons indicating that the piperazine ring is outside the CD cavity, while the aromatic ring system is inside the cavity. Minor changes occur only with γ-CD in the aromatic region. In contrast, the aromatic proton do not show any change with α-CD but the protons in the oxazine ring show a downfield shift which indicates that only the oxazine ring is in the cavity of α-CD. To verify these hypotheses 2D ROESY NMR experiments were carried out with α, β, γ and CMβCD.
2D ROESY NMR results
The 2D ROESY NMR experiment is suitable to obtain information about the spatial proximity between atoms of the host and guest molecules by observing the intermolecular dipolar cross-correlations. The ROE is a manifestation of cross relaxation between two nonequivalent nuclear spins that are relatively close (<5 Å) in space [42].
Figure 7 shows a ROESY spectrum partial expansion of the OFL-CMβCD inclusion complex. Three cross-peaks can be found between the H2, H13 protons of OFL and H3 of CMβCD as well as between H5 of OFL and H5 of CD. These results indicate that OFL enters the CMβCD cavity from the wider rim and the methyl-piperazine ring remains out of the CD cavity. The analogous γ-CD results were the same, the tricyclic moiety is in the cavity. In the case of α-CD the oxazine protons show correlations with the α-CD verifying that only this part of the molecule enters the CD cavity. The molecular modeling study corroborates these 2D NMR results. Figure 8 shows the simplified molecular model of the inclusion of OFL with different CDs.

ROESY spectra of the inclusion complex of OFL with CMβCD using 500 ms mixing time

Molecular structure of OFL—α-CD complex (left) and OFL—β-CD complex (right)
Conclusion
The interactions of OFL with various CDs were characterized by CE, circular dichroism, molecular modeling, and various 1D and 2D NMR techniques. Our results showed that the negatively charged CDs formed the most stable complexes at acidic pH. The best enantioseparation was achieved using CMβCD. 1H NMR chemical shift and 2D ROESY data show that β-CDs and γ-CD accommodate the OFL tricycle in the CD cavity, whereas α-CD lets only in the oxazine ring. Molecular modeling studies provided evidence that the substitution pattern plays an important role in the complex stability. These results offer molecular basis for improved drug formulation aiming especially at targeted delivery.
References
Bolon, M.K.: The newer fluoroquinolones. Med. Clin. N. Am. 95, 793–817 (2011)
Andersson, M.I., MacGowan, A.P.: Development of the quinolones. J. Antimicrob. Chemother. 51, 1–11 (2003)
Ev Lda, S., Schapoval, E.E.: Microbiological assay for determination of ofloxacin injection. J. Pharm. Biomed. Anal. 27, 91–96 (2002)
Fujimoto, T., Mitsuhashi, S.: In vitro antibacterial activity of DR-3355, the S-(−)-isomer of ofloxacin. Chemotherapy 36, 268–276 (1990)
Morrissey, I., Hoshino, K., Sato, K., Yoshida, A., Hayakawa, I., Bures, M.G., Shen, L.L.: Mechanism of differential activities of ofloxacin enantiomers. Antimicrob. Agents Chemother. 40, 1775–1784 (1996)
US Pharmacopaea vol. USP-35-NF-30 (2012)
Bi, W., Tian, M., Row, K.H.: Chiral separation and determination of ofloxacin enantiomers by ionic liquid-assisted ligand-exchange chromatography. Analyst 136, 379–387 (2011)
Zeng, S., Zhong, J., Pan, L., Li, Y.: High-performance liquid chromatography separation and quantitation of ofloxacin enantiomers in rat microsomes. J. Chromatogr. B 728, 151–155 (1999)
Wong, F.A., Juzwin, S.J., Flor, S.C.: Rapid stereospecific high-performance liquid chromatographic determination of levofloxacin in human plasma and urine. J. Pharm. Biomed. Anal. 15, 765–771 (1997)
de Boer, T., Mol, R., de Zeeuw, R.A., de Jong, G.J., Ensing, K.: Enantioseparation of ofloxacin in urine by capillary electrokinetic chromatography using charged cyclodextrins as chiral selectors and assessment of enantioconversion. Electrophoresis 22, 1413–1418 (2001)
Zhou, S., Ouyang, J., Baeyens, W.R., Zhao, H., Yang, Y.: Chiral separation of four fluoroquinolone compounds using capillary electrophoresis with hydroxypropyl-beta-cyclodextrin as chiral selector. J. Chromatogr. A 1130, 296–301 (2006)
Awadallah, B., Schmidt, P.C., Wahl, M.A.: Quantitation of the enantiomers of ofloxacin by capillary electrophoresis in the parts per billion concentration range for in vitro drug absorption studies. J. Chromatogr. A 988, 135–143 (2003)
Horstkotter, C., Blaschke, G.: Stereoselective determination of ofloxacin and its metabolites in human urine by capillary electrophoresis using laser-induced fluorescence detection. J. Chromatogr. B 754, 169–178 (2001)
Szejtli, J.: Cyclodextrin technology. Kluwer Academic, Dordrecht (1988)
Szejtli, J.: Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 98, 1743–1754 (1998)
Sohajda, T., Varga, E., Ivanyi, R., Fejos, I., Szente, L., Noszal, B., Beni, S.: Separation of vinca alkaloid enantiomers by capillary electrophoresis applying cyclodextrin derivatives and characterization of cyclodextrin complexes by nuclear magnetic resonance spectroscopy. J. Pharm. Biomed. Anal. 53, 1258–1266 (2010)
Challa, R., Ahuja, A., Ali, J., Khar, R.K.: Cyclodextrins in drug delivery: an updated review. AAPS PharmSciTech 6, E329–E357 (2005)
Hafner, V., Czock, D., Burhenne, J., Riedel, K.D., Bommer, J., Mikus, G., Machleidt, C., Weinreich, T., Haefeli, W.E.: Pharmacokinetics of sulfobutylether-beta-cyclodextrin and voriconazole in patients with end-stage renal failure during treatment with two hemodialysis systems and hemodiafiltration. Antimicrob. Agents Chemother. 54, 2596–2602 (2010)
Loftsson, T., Duchene, D.: Cyclodextrins and their pharmaceutical applications. Int. J. Pharm. 329, 1–11 (2007)
Buschmann, H.J., Schollmeyer, E.: New textile applications of cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 40, 169–172 (2001)
Koester, L.S., Guterres, S.S., Le Roch, M., Eifler-Lima, V.L., Zuanazzi, J.A., Bassani, V.L.: Ofloxacin/beta-cyclodextrin complexation. Drug Dev. Ind. Pharm. 27, 533–540 (2001)
Jinxia Li, X.Z.: Preparation and characterization of the inclusion complex of ofloxacin with beta-CD and HP-beta-CD. J. Incl. Phenom. Macrocycl. Chem. 69, 173–179 (2011)
Rusu, A., Toth, G., Szocs, L., Kokosi, J., Kraszni, M., Gyeresi, A., Noszal, B.: Triprotic site-specific acid-base equilibria and related properties of fluoroquinolone antibacterials. J. Pharm. Biomed. Anal. 66, 50–57 (2012)
Pop, M.M., Goubitz, K., Borodi, G., Bogdan, M., De Ridder, D.J., Peschar, R., Schenk, H.: Crystal structure of the inclusion complex of beta-cyclodextrin with mefenamic acid from high-resolution synchrotron powder-diffraction data in combination with molecular-mechanics calculations. Acta Crystallogr. B 58, 1036–1043 (2002)
Schmidt, A.K., Cottaz, S., Driguez, H., Schulz, G.E.: Structure of cyclodextrin glycosyltransferase complexed with a derivative of its main product beta-cyclodextrin. Biochemistry 37, 5909–5915 (1998)
Matsumoto, N., Yamada, M., Kurakata, Y., Yoshida, H., Kamitori, S., Nishikawa, A., Tonozuka, T.: Crystal structures of open and closed forms of cyclo/maltodextrin-binding protein. FEBS J. 276, 3008–3019 (2009)
Gyimesi, J., Szökő, É., Magyar, K., Barcza, L.: Determination of drug-cyclodextrin binding constants by capillary zone electrophoresis. J. Incl. Phenom. Macrocycl. Chem. 25, 253–256 (1996)
Rundlett, K.L., Armstrong, D.W.: Methods for the estimation of binding constants by capillary electrophoresis. Electrophoresis 18, 2194–2202 (1997)
Rundlett, K.L., Armstrong, D.W.: Examination of the origin, variation, and proper use of expressions for the estimation of association constants by capillary electrophoresis. J. Chromatogr. A 721, 173–186 (1996)
Shakalisava, Y., Regan, F.: Determination of association constants of inclusion complexes of steroid hormones and cyclodextrins from their electrophoretic mobility. Electrophoresis 27, 3048–3056 (2006)
Chen, Z., Weber, S.G.: Determination of binding constants by affinity capillary electrophoresis, electrospray ionization mass spectrometry and phase-distribution methods. Trends Anal. Chem. 27, 738–748 (2008)
Wren, S.A., Rowe, R.C.: Theoretical aspects of chiral separation in capillary electrophoresis III. Application to beta-blockers. J. Chromatogr. 635, 113–118 (1993)
Liu, X., Lin, H.S., Thenmozhiyal, J.C., Chan, S.Y., Ho, P.C.: Inclusion of acitretin into cyclodextrins: phase solubility, photostability, and physicochemical characterization. J. Pharm. Sci. 92, 2449–2457 (2003)
Mura, P., Bettinetti, G., Melani, F., Manderioli, A.: Interaction between naproxen and chemically modified β-cyclodextrins in the liquid and solid state. Eur. J. Pharm. Sci. 3, 347–355 (1995)
Yap, K.L., Liu, X., Thenmozhiyal, J.C., Ho, P.C.: Characterization of the 13-cis-retinoic acid/cyclodextrin inclusion complexes by phase solubility, photostability, physicochemical and computational analysis. Eur. J. Pharm. Sci. 25, 49–56 (2005)
Schonbeck, C., Westh, P., Madsen, J.C., Larsen, K.L., Stade, L.W., Holm, R.: Hydroxypropyl-substituted beta-cyclodextrins: influence of degree of substitution on the thermodynamics of complexation with tauroconjugated and glycoconjugated bile salts. Langmuir 26, 17949–17957 (2010)
Schonbeck, C., Westh, P., Madsen, J.C., Larsen, K.L., Stade, L.W., Holm, R.: Methylated beta-cyclodextrins: influence of degree and pattern of substitution on the thermodynamics of complexation with tauro- and glyco-conjugated bile salts. Langmuir 27, 5832–5841 (2011)
Allenmark, S.: Induced circular dichroism by chiral molecular interaction. Chirality 15, 409–422 (2003)
Bakirci, H., Zhang, X., Nau, W.M.: Induced circular dichroism and structural assignment of the cyclodextrin inclusion complexes of bicyclic azoalkanes. J. Org. Chem. 70, 39–46 (2005)
Matsuura, N., Takenaka, S., Tokura, N.: Formation of inclusion complexes of benzophenone derivatives: β-cyclodextrin studied by induced circular dichroism. J. Chem. Soc. 2, 1419–1421 (1977)
Harata, K., Uedaira, H.: Circular dichroism spectra of the β-cyclodextrin complex with naphthalene derivatives. Bull. Chem. Soc. Jpn. 48, 375–378 (1975)
Sohajda, T., Beni, S., Varga, E., Ivanyi, R., Racz, A., Szente, L., Noszal, B.: Characterization of aspartame-cyclodextrin complexation. J. Pharm. Biomed. Anal. 50, 737–745 (2009)
Acknowledgments
This work was supported by the National Scientific Research Fund of Hungary, OTKA K73804 and TÁMOP 4.2.1.B-09/1/KMR. This paper was also supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (Sz. B.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tóth, G., Mohácsi, R., Rácz, Á. et al. Equilibrium and structural characterization of ofloxacin–cyclodextrin complexation. J Incl Phenom Macrocycl Chem 77, 291–300 (2013). https://doi.org/10.1007/s10847-012-0245-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-012-0245-2