
Abstract
Four copper(II) complexes containing Schiff base and reduced Schiff base ligands derived from pyridine-2-aldehyde and amino acid containing carboxylate and sulfonate functional groups (N-(2-pyridylmethylene)-amino acid and N-(2-pyridylmethyl)-amino acid, (amino acids = β-alanine and aminoethanesulfonic acid) namely, [Cu(Pbals)(H2O)2]ClO4·H2O 1, [Cu(Pbal)(ClO4)(H2O)] 2, [Cu2(Paes)2(ClO4)2]·2H2O 3, and [Cu(Pae)(H2O)]·ClO4·H2O 4 have been synthesized and characterized. The structural features of carboxylate and sulfonate donor groups have been elucidated. These copper(II) complexes demonstrate different coordination behaviour of the carboxylate and sulfonate groups. Carboxylate groups in complexes 1 and 2 bridge the metal centers and facilitate the formation of 1D helical coordination polymeric structures. In compound 3, the sulfonate groups bridge the metal centers to form a discrete dinuclear complex. In 4, the sulfonate groups link the neighbouring metal centers to form a 1D coordination polymeric structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past decades, extensive research has been devoted in the construction of coordination polymers with diverse topologies. They have potential applications in gas storage, separation, catalysis, magnetism, and sensing [1–5]. Of these, the carboxylate containing ligands have been shown to be excellent for the construction of multidimensional polymers [6–8]. Whereas, studies on sulfonates as building blocks for multidimensional networks are rather scarce in the literature as sulfonate anions are generally considered to be weakly coordinating compared to carboxylates and phosphates. Nonetheless, ligands containing sulfonate groups can be employed for the construction of extended networks with a combination of alkali or alkaline earth metal cations [9, 10]. Several transition and lanthanide complexes containing monodentate and bridging sulfonate groups have been reported in literature [11–13]. In some cases, the sulfonate containing ligands have been utilized in the construction of metal organic frameworks in which the uncoordinated sulfonate groups can functionalize the pore surface for sorption and separation applications [14–16].
As part of our ongoing studies on metal complexes of reduced Schiff base ligands formed between salicylaldehyde and amino acids, we have shown that such ligands can function as tridentate ligands, capable of forming dinuclear unit through bridging phenoxo moiety. Furthermore, the reduced Schiff base ligands are more stable and flexible for coordination upon reduction of their C=N bonds [17, 18]. Previously, we demonstrated that dinuclear Cu(II) complexes of N-(2-hydroxybenzyl)-amino methane/ethane sulfonic acid as both Schiff base and reduced Schiff base ligands form distinct structures and show Catecholase activity. A striking difference between the Schiff base and reduced Schiff base complexes is that the former furnished eight membered ring containing dicopper(II) centers and the sulfonate group acting as a bridging moiety while the later formed the phenoxo bridged Cu2O2 cores [19]. Hence, it was interesting to further investigate the coordination chemistry of the carboxylate and the sulfonate donor group containing Schiff base and reduced Schiff base complexes. Apart from this, amino ethane sulfonic acid which is also known as taurine, a sulfur containing amino acid, is important with respect to various physiological functions. Taurine acts as antioxidant, intracellular osmolyte, membrane stabilizer as well as a neurotransmitter. It may be essential for human infants, and is routinely added to most infant formulas [20]. The studies of complexes of sulphur-containing derivatives have stemmed from their antiviral, anticancer and antibacterial activities.
On the other hand, while N-(2-hydroxybenzyl)-amino acids have been extensively investigated, pyridyl based amino acid derivatives are not well explored. To date, there are only a few publications available on the complexes of N-(2-pyridylmethyl)-amino acid reported by us and others. This ligand system has been shown to generate interesting supramolecular architectures. We have shown that Cu(II) complexes of N-(2-pyridymethyl)-amino acids such as glycine, l-alanine and l-histidine display metallocrown structures [21, 22]. Furthermore, Cu(II), Zn(II), and Pb(II) complexes of N-(2-pyridymethyl)-glycine/alanine exhibit coordination polymeric structures. The influence of anions on the conformation of 1D coordination polymers including zigzag, spiral, and helical systems has been exemplified [23]. Studies by other researchers on similar ligand systems derived from glycine, l-alanine and aminoethanesulfonic acid also disclose fascinating structures, including discrete, [24–27] cubane-like [28, 29] and coordination polymeric structures [30, 31]. Recently, Cu(II) complexes of N-(2-pyridylmethyl)-aspartic acid (H2Pasp) have been found to be helical coordination polymers through carboxylate bridging [32].
Driven by these findings, we were motivated to further study carboxylate- and sulfonate- groups containing Schiff bases and reduced Schiff bases of N-(2-pyridymethyl)-amino acid ligand system. Herein, we report the synthesis, characterization, and solid-state structures of Cu(II) complexes of Schiff base and reduced Schiff base containing pyridyl-based carboxylate and sulfonate groups.
Results and discussion
Synthesis and physical characterization
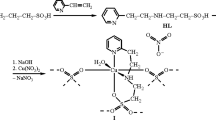
The Schiff base ligands were synthesized by Mannich condensation of pyridine-2-aldehyde, and β-alanine and amino ethane sulfonic acid, respectively, while the reduced Schiff base ligands were obtained by the reduction of the Schiff base with sodium borohydride. Scheme 1 displays the chemical structures of the ligands investigated in this study. The Schiff base ligands were freshly prepared and directly employed for in situ complexation due to their inherent instability while the reduced Schiff base ligands were isolated as their acetic acid salts. The Cu(II) complexes, [Cu(Pbals)(H2O)2]·ClO4·H2O 1, [Cu(Pbal)(ClO4)(H2O)] 2, [Cu2(Paes)2(ClO4)2]·2H2O 3, and [Cu(Pae)(H2O)]·ClO4·H2O 4 were been synthesized and characterized. Slow evaporation or vapor diffusion of the reaction mixture afforded single crystals of compounds 1–3 directly, whereas single crystals of 4, [Cu(pae)(DMF)(H2O)]·ClO4 4a were obtained with a different solution mixture, as described in the experimental section.

Schiff base and reduced Schiff base ligands
In all of these complexes, the IR absorption bands near 3,450 cm−1 indicate the stretching due to water molecules. The C=N stretching frequencies are observed around 1,620 and 1,651 cm−1, respectively, in complexes 1 and 3 indicate characteristic bands due to Schiff base ligand. On the contrary, sharp peaks observed at ~3,230 cm−1 in complexes 2 and 4 are corresponding with the absorption of N–H bond. These results were further confirmed by the reduction of the C=N bond in the ligand. Furthermore, in 1 and 2, the absorption bands around 1,585 and 1,617 cm−1, respectively, correspond to the asymmetric vibrational mode of the carboxylate group [υas(COO−)]. The bands around 1,444 cm−1 are due to the symmetric stretching vibrational mode of the carboxylate group [υs(COO−)]. In this context, the Δυ values observed in complexes 1 and 2 are lower than 200 cm−1, suggesting a bridging mode for the carboxylate group [33]. Whereas, the absorption bands found in 3 and 4 in the range of 1,050–1,386 cm−1 are attributed to the fundamental and split υ3 S–O stretching modes [34].
The UV–Vis spectra for all the complexes showed absorption bands in the range of 692–703 nm and this may be attributed to d–d transitions. Strong absorption bands at 256–289 nm may be due to O → Cu LMCT. The N → Cu LMCT, which are usually around 350 nm were not observed due to the overlapping with the strong and broad O → Cu peak [35]. The solid-state UV–Vis spectra of the complexes do not differ much in solution and in the Nujol mull, suggesting that the coordination and geometry of complexes observed in the solid state are retained in solution. Nonetheless, the LMCT bands are broad in the Nujol mull spectra.
The structural behaviour of all the complexes in methanol solution has been investigated by electrospray ionization mass spectroscopy (ESI–MS). The major peaks of the complexes can be assigned to their Cu:ligand 1:1 species with solvents.
Description of crystal structures
The solid-state structures of complexes 1–4a were determined by X-ray crystallographic techniques. Selected hydrogen bond parameters are given in Table 1. The ligands are coordinated to their Cu(II) centers via using a tridentate mode through the pyridyl, amine/imine and carboxylate/sulfonate groups. It is worthwhile noting that in all of these complexes, the carboxylate and sulfonate bridge the metal centers to form a coordination polymers or a dinuclear cluster.
[Cu(Pbals)(H2O)2]·ClO4·H2O 1
The asymmetric unit of 1 contains a Cu(II) center with a slightly distorted octahedral geometry as illustrated in Fig. 1. The structure is analogous to that of the previously reported [Cu(Pbal)(H2O)2]·ClO4·H2O with differences in the bond distances and the bond angles. In this context, the octahedral geometry around the copper ion is completed by a Schiff base HPbals ligand occupying the equatorial positions in a mer fashion via coordination through pyridyl N (Cu(1)–N(1) = 2.004(2) Å), an amine N (Cu(1)–N(2) = 1.967(2) Å), a carboxylate O (Cu(1)–O(1) = 1.969(2) Å) along with the neighboring carbonyl O (Cu(1)–O(2) = 1.976(2) Å) while two aqua ligands (Cu(1)–O(2S) = 2.639(3) Å and Cu(1)–O(3S) = 2.280(3) Å) occupy the axial sites. In this context, the C=N distance, 1.268(4) Å confirms the integrity of the Schiff base ligand. Furthermore, Cu1 deviates 0.082 Å from the C=N–C plane, confirming the planarity of the Schiff base complex.

A perspective view shows the coordination geometry at Cu(II) and connectivity present in the coordination polymeric cation in 1
The carboxylate group facilitates the formation of a 1D coordination polymer by bridging the Cu(II) centers. The connectivity in the copper center generates a 1D coordination polymer along the b-axis. As expected, the polymeric strands are further supported by hydrogen bonding between aqua ligands and carboxylate O (O2S–H2SA···O1) groups. In this polymeric structure, 1 displays helical conformation in which both left and right handed helices are present in the lattice as shown in Fig. 2a. But the helicity is not well pronounced. Furthermore, both aqua ligands are involved in complementary hydrogen bonding interactions with the adjacent carboxylate, lattice water and perchlorate anion. As shown in Fig. 2b, aqua ligands are linked through lattice water molecules to form a trimeric water cluster. According to the theoretical calculations, the water trimers are more stable in cyclic form with the average O···O distances in the range of 2.799–2.927 Å [36–38]. However, the acyclic water trimer observed here is uncommon [39, 40]. The hydrogen-bonded O···O distances are shorter (O3···O1S, 2.752 Å; O1S···O2S, 2.786 Å) and while O3···O2S distance (3.794 Å) is more the sum of the van der Waals radii [41]. The water trimer is bent in a “L” shape with ∠O3–O1S–O2S angle of 86.5°. These water trimers are also linked to the adjacent coordination polymeric chain as shown in Fig. 2c. Table 1 contains the selected hydrogen bond parameters for 1.

a Left and right handed helical coordination polymeric chains in 1; b polymeric chains in 1 showing the (H2O)3 cluster; c hydrogen bonding interactions of the (H2O)3 cluster with polymeric strands
[Cu(Pbal)(ClO4)(H2O)] 2
Figure 3 displays a ball-and-stick diagram of 2. The deprotonated reduced Schiff base anion Pbal coordinates to the Cu(II) center in a mer-fashion as in 1. The Cu(II) center adopts distorted octahedral geometry in which a pyridyl N (Cu(1)–N(1) = 1.983(3) Å), an amine N (Cu(1)–N(2) = 1.984(3) Å), a carboxylate O (Cu(1)–O(1) = 1.953(2) Å) and a neighboring carbonyl O (Cu(1)–O(2) = 1.955(2) Å) comprise the equatorial plane while an aqua ligand (Cu(1)–O(1S) = 2.765(4) Å) and a perchlorate O (Cu(1)–O(6) = 2.577(6) Å) occupy the axial positions. The C–N distance of 1.472(4) Å and the positioning of Cu(I) away from the C–N–C plane by 1.417 Å confirm the reduction of the C=N bond in the Schiff base ligand and non-planarity of the complex as compared to 1. It is noticeable that despite the non-coordinating nature, the perchlorate is loosely coordinated to the metal center in this case. This differs from the reported Cu(II) complex of HPala in which the perchlorate anion is non-coordinated atom but is involved in weak N–H···O hydrogen bonding and results in a highly ordered behavior of the anion in the crystal lattice [22].

A perspective view showing the coordination geometry of Cu(II) and the connectivity in 2
The metal center is connected to the carbonyl O of neighboring ligand to form a 1D helical coordination polymer along b-axis. Similar to 1, both left and right handed helices are present in the crystal lattice. Figure 4a shows the presence of helicity in the 1D polymeric chain of 2. Along the strand, one of the hydrogen atoms of a aqua ligand is involved in O–H···O bonding with O2. The other hydrogen atom of the aqua ligand and the N–H proton are hydrogen-bonded to an oxygen atom of the perchlorate ligand (Fig. 4b). Since the perchlorate O atoms are disordered, their hydrogen bonding behaviour is not elaborated further here. Table 1 summarizes the hydrogen bond parameters for 2.

a A portion of the 1D coordination polymeric chain of 2. Perchlorate anions are omitted for clarity; b a portion of 2D hydrogen-bonded structure in 2 viewed along the a-axis
In this context, complexes 1 and 2 are similar to each other in terms of their tridentate coordination and crystal packing. Both Λ and Δ helices are present in the crystal lattice in the centrosymmetric space group P21/c. It is noted that both aqua ligands occupy axial positions in 1 while one of the aqua ligands is replaced by a perchlorate anion in 2. This results in a difference in the hydrogen bonding interactions in each complex.
[Cu2(Paes)2(ClO4)2]·2H2O 3
A ball-and-stick diagram of 3 is displayed in Fig. 5. The asymmetric unit of dimeric 3 contains a Cu(II) center with distorted octahedral geometry. At the Cu(II) center, the equatorial plane is occupied by a pyridyl N (Cu(1)–N(1) = 2.016(2) Å), an imine N (Cu(1)–N(2) = 1.969(2) Å), an O from a neighboring sulfonate group (Cu(1)–O(3) = 2.252(1) Å) and an aqua ligand (Cu(1)–O(4) = 1.956(2) Å). The axial positions are occupied by a sulfonate O (Cu(1)–O(1) = 1.986(1) Å) and one of the O atoms from the perchlorate anion (Cu(1)–O(6) = 2.566(2) Å). The bond parameters of the Schiff base ligand demonstrated the characteristic nature in 3, in which the C=N distance is 1.273(3) Å and Cu1 only deviates from the plane of the C=N–C sequence by 0.018 Å.

A ball-and-stick diagram of 3
Though the chain lengths of HPbals and HPaes ligands are comparable, the presence of carboxylate and sulfonate functional groups has resulted in a significant difference in the coordination behavior. Differing from 1 in which the presence of the carboxylate donor group resulted displaying in a 1D coordination polymer in 1, the sulfonate group in 3 gave rise to a discrete dinuclear structure. The dinuclear core forms an interesting eight membered ring consisting of Cu1, O1, S1, O3, Cu1A, O1A, S1A, and O3A with a Cu···Cu non-bonded distance of 4.575 Å. This observation is similar to that reported for the Cu(II) complexes of Schiff base [42] and reduced Schiff base [43] ligands containing phenolate groups.
Complementary hydrogen bonding formation between the hydrogen atoms of the aqua ligand O4, sulfonate O2 and one of the oxygen atoms of the perchlorate anion O8 generated a 2D hydrogen-bonded network in the bc-plane. Figure 6 displays the packing of 3 that shows a portion of the 2D grid formed by the hydrogen-bonded interactions. It can be clearly seen that there are two types of grid with different size and shape. Hydrogen bond parameters for 3 are tabulated in Table 1.

A portion of the 2D grid hydrogen-bonded network in 3
[Cu(Pae)(DMF)(H2O)]·ClO4 4a
A perspective view of 4a is shown in Fig. 7. In this complex, the Pae ligand displays fac-geometry which is different from the other structures 1–3. The Cu(II) center has an octahedral geometry with a pyridyl N (Cu(1)–N(1) = 1.987(3) Å), an amine N (Cu(1)–N(2) = 2.014(3) Å), an O atom of DMF molecule (Cu(1)–O(5) = 1.957(2) Å) and an aqua ligand (Cu(1)–O(4) = 2.026(2) Å) constituting the equatorial plane, while the axial positions are occupied by a sulfonate O (Cu(1)–O(1) = 2.327(2) Å) as well as a sulfonate O from the neighboring strand (Cu(1)–O(3) = 2.467(2) Å). In this case, the DMF molecule is involved in coordination through its amide O atom. In contrast to the planar Schiff base complex 3, here the 4a is non-planar in which Cu1 is deviates 1.526 Å from the C–N–C plane and the C–N distance is 1.480(4) Å.

Ball-and-stick diagram of 4a. The perchlorate anion is omitted for clarity
The sulfonate O bridges the Cu(II) atoms to form a 1D coordination polymer along the b-axis as shown in Fig. 8a. Notably, the oxygen atoms of the perchlorate anions are involved in hydrogen bonding to the N–H proton (N2–H2N···O8), which leads them to be highly ordered in the crystal lattice. Thus, the perchlorates are arranged close to the polymeric strand by intermolecular hydrogen bonds. This observation is quite similar to the reported Cu(II) complex of Pala in which two oxygen atoms of the perchlorate are weakly hydrogen-bonded to the N–H proton [22]. Interestingly, the aqua ligands O4 are hydrogen-bonded to sulfonate O2 atoms of two different 1D coordination polymeric strands as displayed in Fig. 8b. Hence, 4a furnished a 2D hydrogen-bonded ladder structure. In this context, it is noted that a 2D coordination polymer of Cu(II) Pae and azide has been reported recently [31]. Table 1 contains selected hydrogen bond parameters for 4a.

a A portion of the 1D coordination polymer in 4a showing intermolecular interactions; b hydrogen bonds between polymeric chains in 4a
Conclusion
Copper(II) complexes of Schiff base and reduced Schiff base ligands derived from pyridine-2-aldehyde and amino acids have been synthesized and structurally characterized. The influence of carboxylate versus sulfonate groups in Cu(II) complexation has been investigated. In 1 and 2, the tridentate ligand is coordinated in a mer conformation and the carbonyl oxygen in the carboxylate group bridges the neighboring metal centers to form a 1D helical coordination polymers. Both left and right handed helices are present in the lattice. Surprisingly, the coordination of sulfonate ligands in 3 and 4a were found to be entirely different. Complex 3 furnished a eight membered dicopper center with the sulfonate group bridging the Cu(II) atoms whereas 4a possessed a 1D coordination polymeric structure. This is attributed to the fact that the reduction of the rigid C=N bond has resulted in flexibility to the ligand which is likely to be reflected by the construction of a coordination polymer. This study has thus exemplifies the role of sulfonate as well as flexible ligand backbone in the construction of coordination polymers [44].
Experimental
Materials
All chemicals were purchased from Aldrich and used without further purification. Reagents used for the physical measurements were of spectroscopic grade.
Physical measurements
The elemental analyses were performed in the microanalytical laboratory, Department of Chemistry, National University of Singapore. 1H NMR and 13C NMR spectra were recorded on a Bruker ACF 300FT-NMR spectrometer operating in the quadrature mode at 300 MHz. Infrared spectra (KBr pellet) were recorded using an FTS165 Bio-Rad FTIR spectrophotometer in the range of 4,000–400 cm−1. ESI mass spectra were recorded on a Finnigan MAT LCQ mass spectrometer using the syringe pump method. Solvent present in the compounds was determined using an SDT 2960 TGA thermal analyzer with a heating rate of 5 °C min−1 from room temperature to 200 °C in a N2 atmosphere using a 5–10 mg sample per run. The electronic transmittance spectral data were recorded on a Shimadzu UV-2501 PC UV–Vis spectrophotometer in the wavelength of 200–800 nm using Nujol mulls and in MeOH solution.
Synthesis of ligands
All the ligands have been synthesized by Mannich condensation of pyridine-2-aldehyde and the corresponding amino acids following by reduction of the C=N bond. The synthetic procedures of the ligands are similar. Pyridine-2-aldehyde (1.07 g, 10 mmol) in MeOH (10 mL) was added to a solution of the corresponding amino acid (10 mmol) in H2O (10 mL) containing NaOH (0.40 g, 10 mmol). The yellow solution was stirred for 30 min at room temperature prior to cooling in ice bath. NaBH4 (0.45 g, 12 mmol) was added to reduce the intermediate Schiff base. The solution was stirred for another 1 h before acidified with acetic acid to pH 4. Then the solvent was evaporated to dryness and methanol was added to extract the product. Excess of Et2O was added to get the yellowish precipitate. The precipitate was filtered under nitrogen and dried under vacuum. Owing to the hygroscopic nature, isolation of analytical pure ligand was not possible in our hands, therefore exact yield and consistent elemental analysis results could not be obtained. Nonetheless, all the ligands have been isolated as acetic acid salts and characterized by 1H NMR and 13C NMR to elucidate the chemical structure.
N-(2-pyridylmethyl)-β-alanine (HPbal)
1H NMR (D2O, ppm): δ 8.58 (d, 1H, Py), 7.91 (dt, 2H, Py), 7.45–7.54 (m, 2H, Py), 4.39 (s, 2H, –CH2NH), 3.30 (t, 2H, –NHCH2), 2.61 (t, 2H, –CH2COOH). 13C NMR (D2O, ppm): δ 180.61 (–COOH), 152.84, 152.1, 141.20, 127.19, 126.78 (Py), 53.63 (–CH2NH), 47.00 (–CH2NH), 34.94 (–CH2COOH).
N-(2-pyridylmethyl)-amino ethane sulfonic acid (Hpae)
1H NMR (D2O, ppm): δ 8.63 (d, 1H, Py), 7.95 (dt, 2H, Py), 7.49–7.57 (m, 2H, Py), 4.45 (s, 2H, –CH2NH), 3.56 (t, 2H, –CH2SO3H), 3.35 (t, 2H, –NHCH2). 13C NMR (D2O, ppm): δ 153.15, 152.05, 141.19, 127.08, 126.62 (Py), 54.06 (–CH2NH), 49.54 (–CH2SO3H), 45.65 (–CH2NH).
Synthesis of complexes
Caution ! Perchlorate salts are potentially explosive. Care must be exercised while handling although we have not encountered any unpleasant situation.
[Cu(Pbals)(H2O)2]ClO4·H2O 1
Pyridine-2-aldehyde (0.054 g, 0.5 mmol) in MeOH (2 mL) was added to a solution of β-alanine (0.045 g, 0.5 mmol) in H2O (2 mL) containing NaOH (0.02 g, 0.5 mmol). The yellow solution was stirred for 30 min. Copper perchlorate hexahydrate (0.185 g, 0.5 mmol) in MeOH (2 mL) was added to the solution to facilitate in situ complexation. The mixture was stirred for 30 min and filtered. The block-like blue crystals were obtained from the filtrate and then dried in air. Yield: 0.13 g, (74%). Anal. Calcd. for C9H11N2O7ClCu (358.19): C, 30.18; H, 3.10; N, 7.82; found: C, 30.66; H, 3.18; N, 7.57%. IR (KBr, cm−1): υ(OH) 3455; υ(C=N) 1620; υas(COO−) 1585; υas(COO−) 1443. Calcd. TG weight loss for 1 H2O 4.8%; found 4.9%. ESI–MS [Cu(Pbals)(H2O)2]+ 271.8. UV–Vis (λmax (nm), ε(M−1 cm−1)): (methanol) LMCT, 289(1550); d–d transition, 703(90); (Nujol mull) LMCT, 679; d–d transition, 295.
[Cu(Pbal)(ClO4)(H2O)] 2
To a solution of Hpbal (0.09 g, 0.5 mmol) in MeOH (5 mL), Cu(ClO4)2·6H2O (0.185 g, 0.5 mmol) in MeOH (5 mL) was added. The mixture was stirred for 30 min and filtered. Block-like blue crystals were obtained from the filtrate by slow evaporation. Yield: 0.05 g, (25%). Anal. Calcd for C9H13N2O8ClCu (376.21): C, 28.73; H, 3.48; N, 7.45; found: C, 28.31; H, 3.52; N, 7.30%. IR (KBr, cm−1): υ(OH) 3518; υ(N–H) 3249; υas(COO−) 1617; υs(COO−) 1445. Calcd. TG weight loss for 1 H2O 5.0%; found 5.0%. ESI–MS [Cu(Pbal)(CH3OH)]+ 273.7. UV–Vis (λmax (nm), ε(M−1 cm−1)): (methanol) LMCT, 256(4760); d–d transition, 693(690); (Nujol mull) LMCT, 723; d–d transition, 251.
[Cu2(Paes)2(ClO4)2]·2H2O 3
Pyridine-2-aldehyde (0.054 g, 0.5 mmol) in MeOH (2 mL) was added to a solution of amino ethane sulfonic acid (0.063 g, 0.5 mmol) in H2O (2 mL) containing NaOH (0.02 g, 0.5 mmol). The yellow solution was stirred for 30 min. Copper perchlorate hexahydrate (0.185 g, 0.5 mmol) in MeOH (2 mL) was added to the solution to facilitate in situ complexation. The mixture was stirred for 30 min and filtered. Vapor diffusion of Et2O into the filtrate afforded rod-like green crystals. Yield: 0.14 g, (35%). Anal. Calcd. for C16H22N4S2O16Cl2Cu2 (788.50): C, 24.37; H, 2.81; N, 7.11; S, 8.13; found: C, 24.18; H, 2.62; N, 6.91; S, 7.97%. IR (KBr, cm−1): υ(OH) 3470; υ(C=N) 1651; υ(SO3 −) 1386, 1072. Calcd. TG weight loss for 2 H2O 4.6%; found 4.9%. ESI–MS [Cu(Paes)(CH3OH)]+ 307.8. UV–Vis (λmax (nm), ε(M−1 cm−1)): (methanol) LMCT, 287(16210); d–d transition, 692(290); (Nujol mull) LMCT, 704; d–d transition, 292.
[Cu(Pae)(H2O)]ClO4·H2O 4
To a solution of Hpae (0.11 g, 0.5 mmol) in MeOH (3 mL), Cu(ClO4)2·6H2O (0.185 g, 0.5 mmol) in MeOH (3 mL) was added. The blue precipitate obtained after stirring for 1 h was filtered off, wash with MeOH (2 mL), Et2O (10 mL) and then dried under vacuum. Yield: 0.16 g, (70%). Anal. Calcd. for C8H15N2S2O9ClCu (414.28): C, 23.19; H, 3.65; N, 6.76; S, 7.74; found: C, 23.92; H, 3.39; N, 6.85; S, 7.50%. IR (KBr, cm−1): υ(OH) 3449; υ(NH) 3227; υ(SO3 −) 1233, 1178, 1050. Calcd. TG weight loss for 2 H2O 8.7%; found 8.4%. ESI–MS [Cu(Pae)(CH3OH)]+ 309.6. UV–Vis (λmax (nm), ε(M−1 cm−1)): (methanol) LMCT, 257(5920); d–d transition, 700(55); (Nujol mull) LMCT, 254; d–d transition, 692.
Single crystal of 4 were obtained as [Cu(pae)(DMF)(H2O)]·ClO4 4a by slow evaporation of the reaction mixture from methanol and DMF. Anal. Calcd. for C11H18N3SO8ClCu (469.36): C, 28.15; H, 4.29; N, 8.95; S, 6.33; found: C, 27.93; H, 4.32; N, 8.76; S, 6.88.
X-ray crystallography
Single crystals X-ray diffraction measurements were carried out on a Bruker AXS APEX CCD diffractometer. Unit cell dimensions were obtained with least-squares refinements, and all structures were solved by direct methods. The program SMART was used for collecting frames of data, indexing reflections and determination of lattice parameters. SAINT [45] for integration of intensity of reflections and scaling; SADABS [46] was used for empirical absorption correction and SHELXTL [47] was used for space group determination, structure solution and least-squares refinements on F 2. Selected crystallographic data and refinement details are displayed in Table 2. All the C–H hydrogen atoms were placed in their appropriate calculated positions using riding models. All the non-hydrogen atoms except the oxygen of the perchlorate were refined anisotropically. Soft constraint option SADI was used to make Cl–O distances in the perchlorate anions equal. All the hydrogen atoms of the water molecules were located. In complexes 1, 3 and 4a, all the oxygen atoms of perchlorate anions were resolved with full occupancy. In 3, the disordered oxygen atoms in perchlorate anion were resolved in 0.5 occupancy.
Supplementary material
CCDC 825096–825099 contain the supplementary crystallographic data in CIF format for 1–4a. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (international) +44-1223/336-033; e-mail: deposit@ccdc.cam.ac.uk].
References
Moulton, B., Zaworotko, M.J.: From molecules to crystal engineeering. Supramolecular isomerism and polymorphism in network solids. Chem. Rev. 101, 1629–1658 (2001)
Kitagawa, S., Kitaura, R., Noro, S.-I.: Functional porous coordination polymers. Angew. Chem. Int. Ed. 43, 2334–2375 (2004)
Rowsell, J.L.C., Yaghi, O.M.: Strategies for hydrogen storage in metal-organic frameworks. Angew. Chem. Int. Ed. 44, 4670–4679 (2005)
Biradha, K., Sarkar, M., Rajput, L.: Crystal engineering of coordination polymers using 4,4′-bipyridine as a bond between transition metal atoms. Chem. Commun. 40, 4169–4179 (2006)
Robin, A.Y., Fromm, K.M.: Coordination polymer networks with O- and N-donors: What they are, why and how they are made. Coord. Chem. Rev. 250, 2127–2157 (2006)
Ye, B.-H., Tong, M.-L., Chen, X.-M.: Metal-organic molecular architectures with 2, 2′-bipyridyl-like and carboxylate ligands. Coord. Chem. Rev. 249, 545–565 (2005)
Eddaoudi, M., Moler, D.B., Li, H., Chen, B., Reineke, T.M., O’Keeffe, M., Yaghi, O.M.: Modular chemistry: Secondary building units as a basis for the design of highly porous and robust metal-organic carboxylate frameworks. Acc. Chem. Res. 34, 319–330 (2001)
Rao, C.N.R., Natarajan, S., Vaidhyanathan, R.: Metal carboxylates with open architectures. Angew. Chem. Int. Ed. 43, 1466–1496 (2004)
Coté, A.P., Shimizu, G.K.H.: The supramolecular chemistry of the sulfonate group in extended solids. Coord. Chem. Rev. 245, 49–64 (2003)
Videnova-Adrabinska, V.: Coordination and supramolecular network entanglements of organodisulfonates. Coord. Chem. Rev. 251, 1987–2016 (2007)
Yang, X., Rivers, J.H., McCarty, W.J., Wiester, M., Jones, R.A.: Influence of metal-ligand ratio on benzimidazole based luminescent lanthanide complexes: 3-D network structures and chloride anion binding. New J. Chem. 32, 790–793 (2008)
Cai, J., Chen, C.-H., Feng, X.-L., Liao, C.-Z., Chen, X.-M.: A novel supramolecular synthon for H-bonded coordination networks: syntheses and structures of extended 2-dimensional cadmium(II) arenedisulfonates. Dalton Trans. 2370–2375 (2001). doi:10.1039/B102729H
Cai, J., Chen, C.-H., Liao, C.-Z., Yao, J.-H., Hu, X.-P., Chen, X.-M.: Variation in the coordination mode of arenedisulfonates to copper(II): synthesis and structural characterization of six copper(II) arenedisulfonate complexes. Dalton Trans. 1137–1142 (2001). doi: 10.1039/B009851P
Horike, S., Bureekaew, S., Kitagawa, S.: Coordination pillared-layer type compounds having pore surface functionalization by anionic sulfonate groups. Chem. Commun. 471–473 (2008). doi:10.1039/B715481J
Gao, Q., Wu, M.-Y., Huang, Y.-G., Chen, L., Wei, W., Zhang, Q.-F., Jiang, F.-L., Hong, M.-C.: Double-walled tubular metal-organic frameworks constructed from bi-strand helices. Cryst. Eng. Commun. 11, 1831–1833 (2009)
Mi, L., Hou, H., Song, Z., Han, H., Fan, Y.: Polymeric zinc rerrocenyl sulfonate as a molecular aspirator for the removal of toxic metal ions. Chem. Eur. J. 14, 1814–1821 (2008)
Ganguly,R., Sreenivasulu,B., Vittal, J.J.: Amino acid containing reduced Schiff base as the building blocks for supramolecular structures. Coord. Chem. Rev. 252, 1027–1050 (2008)
Vittal, J.J.: Hydrogen-bnded coordination polymeric structures. In: Tiekink, E.R.T., Vittal, J.J. (eds.) Frontiers in Crystal Engineering, pp. 297–319. Wiley, Chichester (2006)
Sreenivasulu, B., Vetrichelvan, M., Zhao, F., Gao, S., Vittal, J.J.: Copper(II) complexes of Schiff base and reduced Schiff base ligands: influence of weakly coordinating sulfonate groups on the structure and oxidation of 3,5-DTBC. Eur. J. Inorg. Chem. 2005, 4635–4645 (2005)
Huxtable, R.J.: Physiological actions of taurine. Physiol. Rev. 72, 101–163 (1992)
Vittal, J.J., Wang, X., Ranford, J.D.: Influence of the Li+ on the structure of [Cu3(phis)3]3+ cation. Inorg. Chem. 42, 3390–3392 (2003)
Wang, X., Vittal, J.J.: Nature of the reactants and influence of water on the supramolecular assembly. Inorg. Chem. 42, 5135–5142 (2003)
Wang, X., Ranford, J.D., Vittal, J.J.: One dimensional coordination polymers: Cu(II) and Zn(II) complexes of N-(2-pyridylmethyl)-glycine and N-(2-pyridylmethyl)-l-alanine. J. Mol. Struct. 796, 28–35 (2006)
Correia, V.R., Bortoluzzi, A.J., Neves, A., Joussef, A.C., Vieira, M.G.M., Batista, S.C.: Bis[N-(2-pyridylmethyl)glycinato]zinc(II) dihydrate. Acta. Crystallogr. E. 59, m464–m466 (2003)
Li, J.-X., Jiang, Y.-M., Li, H.-Y.: Bis[2-(2-pyridylmethylamino)ethanesulfonato-kappa(3) N, N′, O]cobalt(II). Acta. Cryst. E62, m2984–m2986 (2006)
Liao, B.-L., Li, J.-X., Jiang, Y.-M.: Bis[2-(2-pyridylmethylamino)ethanesulfonato-κ N 3,N 1,O]nickel(II). Acta. Cryst. E63, m1974–u1674 (2007)
Li, J.-X., Jiang, Y.-M.: Lian, B.-R.: Bridging chlorides and hydroxides: syntheses and crystal structures of binuclear and tetranuclear Ni-II complexes derived from a reduced Schiff-base ligand 2-(2-pyridylmethylamino)ethanesulfonic acid. J. Chem. Crystallogr. 38, 711–715 (2008)
Ama, T., Okamoto, K.-i., Yonemura, T., Kawaguchi, H., Takeuchi, A., Yasui, T.: Tetranuclear cobalt(III) complex having the cubane Co4O4 core: synthesis and structural analysis of the complex containing (2-Pyridylmethyl)glycine. Chem. Lett. 11, 1189–1190 (1997)
Li, J.-X., Jiang, Y.-M., Chen, M.-J.: Bridging chlorides and hydroxides: syntheses and crystal structures of binuclear and tetranuclear Ni-II complexes derived from a reduced Schiff-base ligand 2-(2-pyridylmethylamino)ethanesulfonic acid. J. Coord. Chem. 61, 1765–1773 (2008)
Li, J.-X., Jiang, Y.-M., Wang, Y.-F., Liang, D.-W.: Di-μ-azido-κ- N4:N-bis{aqua[2-(2-pyridylmethylideneamino)ethanesulfonato-κ N3, N1, O]nickel(II)} dihydrate. Acta. Cryst. E. 61, m160–m162 (2005)
Li, J.-X., Jiang, Y.-M., Wang, Y.-F.: Poly[μ-azido-κN2(1): N3-μ-2-(2-pyridylmethylamino)ethanesulfonato-κN4, N′, O: O′-copper(II)]. Acta. Cryst. E. 63, m213–m215 (2007)
Wu, S.-P., Lee, C.-H.: Infinite chiral single-helical structures formed by the self-assembly of Cu(II)-N-(2-pyridylmethyl)-aspartate complexes. Cryst. Eng. Commun. 11, 219–222 (2009)
Nakamoto, K.: Infrared and Raman spectra of inorganic and coordination compounds. Wiley, New York (1986)
Cai, J.W., Chen, C.-H., Liao, C.-Z., Yao, J.-H., Hu, X.-P., Chen, X.-M.: Variation in the coordination mode of arenedisulfonates to copper(II): synthesis and structural characterization of six copper(II) arenedisulfonate complexes. J. Chem. Soc. Dalton Trans. 7, 1137–1142 (2001)
Lever, A.B.P.: Inorganic electronic spectroscopy. Elsevier, Amsterdam (1984)
Ludwig, R.: Water: from clusters to the bulk. Angew. Chem. Int. Ed. 40, 1808–1827 (2001)
Mas, E.M., Bukowski, R., Szalewicz, K.: Ab initio three-body interactions for water. I. potential and structure of water trimer. J. Chem. Phys. 118, 4386–4403 (2003)
Ghosh, S.K., Ribas, J., Bharadwaj, P.K.: Characterization of 3-D metal-organic frameworks formed through hydrogen bonding interactions of 2-D networks with rectangular voids by CoII and NiIIpyridine-2, 6-dicarboxylate and 4, 4′-bipyridine or 1, 2-di(pyridyl)ethylene. Cryst. Growth Des. 5, 623–629 (2005)
Ghosh, S.K., Ribas, J., Bharadwaj, P.K.: Metal-organic framework structures of Cu(II) with pyridine-2, 6-dicarboxylate and different spacers: identification of a metal bound acyclic water tetramer. Cryst. Eng. Commun. 6, 250–256 (2004)
Ghosh, S.K., Bharadwaj, P.K.: Metal-organic framework H-bonded like a polycatenane: coexistence of acyclic water trimer and nonomer. Inorg. Chem. 44, 5553–5555 (2005)
Bondi, A.: van der Waals volumes + radii. J. Phys. Chem. 68, 441 (1964)
Sreenivasulu, B., Zhao, F., Gao, S., Vittal, J.J.: Synthesis, structures and catecholase activity of new series of dicopper(II) complexes of reduced Schiff base ligands. Eur. J. Inorg. Chem. 2006, 2656–2670 (2006)
Li, J.-X., Jiang, Y.-M., Wang, J.-G.: Catena-Poly[[(thiocyanato-kappa N)copper(II)]μ3-[2-(2-pyridylmethylamino)ethanesulfonato-κ N5, N′, O : O′: O′′]]. Acta. Cryst. E63, m601–m603 (2007)
Leong, W.L., Vittal, J.J.: One-dimensional coordination polymers: complexity and diversity in structures, properties, and applications. Chem. Rev. 111, 688–764 (2011)
SMART & SAINT: Software reference manuals, version 5.6. Bruker Analytical X-ray Systems, Inc., Madison (2003)
Sheldrick, G.M.: SADABS a software for empirical absorption correction. University of Göttingen, Göttingen (2003)
SHEXTL: Reference manual, version 6.14. Bruker Analytical X-ray Systems, Inc., Madison (2003)
Acknowledgments
We thank the Ministry of Education, Singapore, for the financial support through the National University of Singapore (Grant No. R-143–000-439–112). Ms. G. K. Tan and Prof. L. L. Koh of the X-ray facility are thanked for their help in X-ray crystallography.
Author information
Authors and Affiliations
Corresponding author
Additional information
This is dedicated to Professor Leonard F. Lindoy for his contributions to chemistry.
Rights and permissions
About this article
Cite this article
Leong, W.L., Vittal, J.J. Synthesis, characterization and structures of copper(II) complexes containing carboxylate and sulfonate groups. J Incl Phenom Macrocycl Chem 71, 557–566 (2011). https://doi.org/10.1007/s10847-011-0015-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-011-0015-6