
Abstract
Co-doped ceria of Ce0.8Gd0.2−x Ca x O2-δ (x = 0−0.2), were prepared by oxalate co-precipitation method. Their structures and conductivities were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and AC impedance spectroscopy (IS). All the electrolytes were found to be single phase with cubic fluorite structure. SEM cross-section image showed relatively uniform grains with distinct and clean grain boundaries. The chemical states of the surface of the prepared samples were analyzed by XPS. Though Gd and Ca were present in their characteristic chemical state, Ce was found in single Ce4+ state or in mixed Ce4+ and Ce3+ states. IS measurements indicated that the conductivities for Ce0.8Gd0.2–x Ca x O2-δ pellets increased with increasing the sintering temperature. Moreover, co-doping with appropriate ratio of gadolinium and calcium was found to effectively enhance the conductivity in comparison to the singly doped ceria. The isothermal conductivity plots showed that sample Ce0.8Gd0.1Ca0.1O2-δ had the maximum conductivity with minimum activation energy (σ 700°C = 0.0742 S/cm, Ea = 0.58 eV), which is much higher compared to the conductivity exhibited by most of the reported codoped ceria compositions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the past two decades researches on solid oxide fuel cells (SOFCs) have attracted worldwide interests due to their high efficiency and environmental benefits [1]. The conventional electrolytes employed in SOFCs are yttria-stabilized zirconia (YSZ) because of its nearly pure oxygen ionic conductivity in an oxidizing or a reducing environments and good mechanical properties. However, high operation temperatures above 1000°C are required for this electrolyte due to its relatively low ionic conductivity. Such high temperatures increase the fabrication cost and accelerates degradation of the fuel cell system. Decreasing the operating temperature is, therefore, being attracted great attentions for the long-term performance stability and lowering cost of SOFCs [2]. However, decreasing operation temperature requires increasing electrolyte ionic conductivity and enhancing electrode reaction activity. In the absence of available electrolyte materials at low temperatures, maximizing electrolyte ionic conductivity is of significant importance. As a result, enormous amounts of effort on ionic conductivity improvement are found in the literatures for the oxide electrolyte materials, including ceria-based oxides, lanthanum gallate-based oxides, and Ln10-x Si6O26-based apatites [3–6].
Doped-ceria fluorites have been considered as one of the most promising electrolytes for intermediate temperature solid oxide fuel cells (IT-SOFCs) due to their higher oxygen ionic conductivity and their good chemical compatibility with ferritic stainless steels [7]. The most effective dopants which enhance the ionic conductivity of the ceria electrolyte were found to be Gd2O3 and Sm2O3 [4]. Recently, another co-doping strategy to further improve the ionic conductivity of ceria-based electrolytes was developed [8–17]. For example, Yamamara et al. [16] found that the electrical conductivity in the Ce1-x-y La x M y O2-δ (M=Ca, Sr) system showed multiple doping effects due to suppressing the ordering of oxygen vacancy. Banerjee et al. [17] recently reported a novel series of Ce0.8Sm0.2–x Ca x O2–δ compositions and found that they exhibited a significant improvement in properties compared to the single-doped Ce0.8Sm0.2O1.9. All the results showed that co-doping ceria with alkaline earth and rare earth ion exhibited higher conductivity than the singly doped ceria.
In the present study, a series of co-doped Ce0.8Gd0.2−x Ca x O2-δ (x = 0–0.2) compositions were prepared by oxalate co-precipitation method to further verify the effect of calcium co-addition on the electrical properties of these ceramics. Calcium was chosen as a co-dopant primarily due to its stable valence and suitable cationic radius, along with its improvement effect on the ionic conductivity exhibited by the ceria–samarium solid electrolyte [17, 18].
2 Experimental
2.1 Sample preparation
An oxalate co-precipitation method was used to prepare the precursor powders of Ce0.8Gd0.2−x Ca x O2-δ ceramics. The appropriate quantities of Ce(NO3)3·6H2O (99.99%), Gd(NO3)3·5H2O (99.99%) and Ca(NO3)2·4H2O (99.9%) were dissolved separately in water, mixed and co-precipitated with ammonium oxalate solution. The precipitant was dried after filtration and rinsing. The dried powders were calcined at 400, 600, 700 and 800°C for 4 h in air. The calcined powders were uniaxially pressed (10 MPa) into powder compacts and subsequently isostatically pressed at 180 MPa pressure. The green bodies were sintered at 1200–1400°C for 2 h. The relative density of the sintered samples was measured by Archimedes principle.
2.2 Property measurements
Thermal decomposition of the precursors was monitored by Thermogravimetric and Differential Scanning Calorimeter (TG-DSC) with instrument NETZSCH STA 409 PC/PG in flowing air with heating rate of 10°C/min in alumina crucible up to 800°C. The elemental analysis was performed with a Flash EA 1112 elemental analyzer.
Powder X-ray diffraction (XRD) intensities were recorded with X-ray diffractometer (PANalytical X’ pert PRO, Netherlands) operating at 40 kV/40 mA, using CuKα radiation. Data in the angular region of 2θ = 10–80° were collected in a step-scanning mode, with a step length of 0.0167° and a step-counting time of 5.08 s. A cerium oxide sample was used as an instrument standard to allow for the instrument broadening correction. The average crystallite size D of the calcined powders was calculated using the (111) peak of the fluorite structure by the Scherrer formula D = 0.9λ/βcosθ, where λ is the wavelength of the X-ray (1.5418 Å), θ the diffraction angle and β describes the structural broadening, which is the difference in integral profile width between the standard and the unknown sample. The integral profile width of the CeO2 standard sample was determined to be 0.098°. The crystalline phase was analyzed with PANalytical X’Pert High Score Plus program and identified using the International Centre for Diffraction Data (ICDD) Powder Diffraction Files (PDF). Lattice parameters were calculated by Rietveld method.
X-ray photoelectron spectroscopy (XPS) measurements were carried out at room temperature on a Perkin Elmer PHI 5300 X-ray Photoelectron Spectrometer. The angle between the X-ray beam and the analyzer was 80° and the photoelectrons were collected at an angle of 45°. Al Ka radiation (hv = 1486.6 eV) was adopted as the excitation source, operating at 250 W with 12.5 kV acceleration voltage. The pass energy of the survey scan was 89.45 eV and that of detail scan was 35.75 eV with a resolution of 1 and 0.2 eV, respectively. Before being sent to the chamber, each sample was cleaned carefully. The base pressure of the instrument was 9.5 × 10−8 Torr (1 Torr = 133.3 Pa). The binding energies (BE) were corrected using the background C1s peak (284.6 eV) as a reference. The peak position was estimated using a fitting procedure based on summation of Lorentzian and Gaussian functions using the XPSPEAK 4.1 program [19].
The size distribution and shape of the calcined powder was observed using transmission electron microscopy (TEM, Tecnai G2 20, FEI Company, Netherlands). The microstructures of the sintered pellets were revealed by scanning electron microscopy (SEM, JSM-6700F).
2.3 Electrical measurements
The ionic conductivity measurements were performed by an ac (alternating current) complex impedance method at frequencies range from 0.1 Hz to 1 MHz on a PARSTAT 2273 impedance analyzer. Before measurements, Platinum paste was painted on two sides of the disk as electrodes, and the disk was sintered at 1000°C for 10 min to erase the solvent. The measurements were conducted in air in the temperature range from 250 to 800°C with an increment of 50°C. Curve fitting and resistance calculation were done by ZSimpWin software. The conductivities were calculated using the expression of σ = l/SR, where l is the sample thickness and S is the electrode area of the sample surface. Activation energies (E a) were calculated by fitting the conductivity data to the Arrhenius relation for thermally activated conduction, which is given as σT = σ 0 exp(-E a /kT), where σ, σ 0 , E a , k, T are the conductivity, pre-exponential factor, activation energy, Boltzmann constant and absolute temperature, respectively.
3 Results and discussion
3.1 Powder characterization
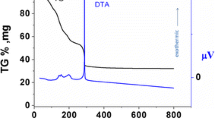
In an oxalate co-precipitation method employing ammonium oxalate as the precipitant, the composition of the as–dried precursor was detected from XRD patterns. The composition of a precursor representing Ce0.8Gd0.15Ca0.05O2–δ sample was analyzed and found to be composed of Ce2(C2O4)3·10H2O (PDF No 00-020-0268), Gd2(C2O4)3·10H2O (PDF No 00-020-0411) and CaC2O4·H2O (PDF No 00-014-0771) (Fig. 1). To further determine the ingredient, elemental analysis (EA) was used and the results showed that the precursor contained 10.46 wt.% of C and 1.69 wt.% of H. N was not detected positively (detection limit: 0.01 wt.%), indicating it was non-precipitating. In addition, thermal behavior of the precursor was investigated and shown in Fig. 2. As can be seen, the decomposition of the precursor proceeds through two distinct stages. The first stage with 22.5% mass loss and two endothermic peaks at 110 and 168°C may be attributed to the evaporation of water of crystallization. The second stage with 30.6% weight loss and exothermic peak at 332°C can be ascribed to the decomposition of the oxalate precursor. No remarkable weight loss could be detected above 400°C, suggesting the formation of pure phase, as confirmed in Fig. 3.

XRD patterns of the as–dried oxalate co-precipitate and the referenced PDF index species

TG-DSC curves of co-precipitated powder conducted at a heating rate of 10°C/min to 800°C in air

XRD patterns of the Ce0.8Gd0.15Ca0.05O2-δ powders calcined for 4 h at different temperature
Based on the results of XRD, EA and TG/DSC, the composition of the precursor was determined, and more importantly, it can be deduced that the added metal ions co-precipitated completely in the light of the nominal composition.
Figure 3 shows the X-ray diffraction patterns obtained for the precursor of Ce0.8Gd0.15Ca0.05O2–δ specimen calcined at 400, 600, 700 and 800°C. It can be seen that all the samples have a cubic fluorite structure, which indicates that by the oxalate pyrogenation route, relatively low calcination temperature is needed to form the fluorite phase. It is noteworthy that the XRD peaks at 400°C are broad, suggesting the presence of nano-scale particles. However, the peaks gradually sharpen with increasing the temperature. The crystal sizes of the powders calculated with Scherrer’s equation are 14.2 nm, 22.5 nm and 38.1 nm for Ce0.8Gd0.15Ca0.05O2–δ calcined at 400°C, 600°C and 800°C for 4 h, respectively. The morphology of the sample calcined at 600°C was observed by transmission electron microscopy (TEM). The particles were found to be softly agglomerated with particle size ~22 nm (Fig. 4), which is in accordance with that estimated size based on the Scherrer equation.

TEM micrograph of Ce0.8Gd0.15Ca0.05O2-δ powder calcined at 600°C for 4 h
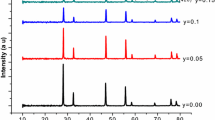
The XRD patterns of the Ce0.8Gd0.2−x Ca x O2-δ (x = 0–0.2) powders calcined at 600°C for 4 h are displayed in Fig. 5. A qualitative analysis of the XRD data indicated that all samples exhibited a cubic phase (fluorite type crystal structure, space group F m-3m ), as expected. The structural study was performed using the Rietveld analysis program, X’Pert High Score Plus. The F m-3m space group was assumed with (Gd3+, Ca2+, Ce4+) cations and O2− anions in 4a and 8c positions, respectively [20]. The lattice parameter, a, was refined and listed in Table 1. It can be seen that the cell parameters increase with x (calcium concentration) increasing in the investigated substitution range of x = 0–0.20. According to the elastic energy, Kim’s expression implies that the effective ionic radius of ceria doping with trivalent and divalent metals is 0.1038 and 0.1106 nm, respectively [7]. Since the radius of Gd3+ (0.1053 nm) and Ca2+ (0.112 nm) [21] are larger than the critical radius, the cell parameter increases with increasing Ca2+ content, indicating that Gd and Ca are well into the crystal lattice of ceria.

XRD patterns of Ce0.8Gd0.2−x Ca x O2-δ calcined at 600°C for 4 h
3.2 Sintering behavior and microstructure
The relative densities of the typical Ce0.8Gd0.1Ca0.1O2−δ samples sintered at 1200, 1300 and 1400°C for 2 h are measured to be 92.5, 95.3 and 96.6%, respectively. The effect of sintering temperature on density and microstructure of the co-precipitated powders was further characterized by SEM. Figure 6 shows the microstructural evolution of Ce0.8Gd0.1Ca0.1O2−δ samples in the temperature of 1200–1400°C. As illustrated in Fig. 6(a–c), some micropores are present but the number of the micropores is gradually reducing with increasing temperature. On the other hand, it is observed that the sample has relatively uniform grain size distribution and the grain size increases from 0.5–1 μm to 1–2 μm. This is consistent with the results of the relative density measurements. Figure 6(d) is the SEM cross-section image of Ce0.8Gd0.1Ca0.1O2−δ pellet sintered at 1400°C. It can be seen that the sample has uniform grain size and the grains are connected to each other compactly. Furthermore, from the distinct and clean grain boundaries in the image, we can conclude that Gd and Ca additives distribute uniformly in the sample.

SEM images of Ce0.8Gd0.10Ca0.10O2-δ sintered at 1200°C (a), 1300°C (b), 1400°C (c) and cross-section image sintered at 1400°C (d)
3.3 Electrical conductivity
To better know the effect of Ca additions on the electrical response of the Gd-doped ceria samples, the electrical measurements were carried out on those sintered at 1400°C with a sufficiently high relative density (96.6%). Figures 7, 8 and 9 show the impedance measurements for Ce0.8Gd0.2−x Ca x O2-δ measured at 250, 450, and 700°C, respectively. It can be seen from Fig. 7 that the impedance plot consists of a depressed arc at high and middle frequencies that is dependent on the Ca content. As the temperature increased, the time constants of the relaxations resulting from the individual polarizations are reduced and the arcs are shifted to higher frequencies (Fig. 8). At higher temperatures, the depressed arcs are not observed and the spike collapses to a semicircular arc, indicating that oxygen-ions are able to diffuse through the entire thickness of the electrode (Fig. 9). It is noteworthy that there emerges a large “tail” in the high-frequency region in Fig. 9. This may be attributed to the inductance of the experiment [22].

Typical plot of the IS measurements at 250°C

Typical plot of the IS measurements at 450°C

Typical plot of the IS measurements at 700°C
The characteristic frequencies of the different processes are shown by arrows in Figures 7, 8 and 9. As expected, the frequencies shifted to the higher side with increasing temperature. From Fig. 7, the capacitance (C) values were calculated using the equation 2πfRC = 1 (f is frequency) [23]. It was found that the capacitance values were in the range usually expected (10−10–10−7F) for resistive grain-boundary processes. The capacitance of the grain-relaxation process is much smaller compared to that of the grain boundary and is typically in the range of picofarads [23]. Hence, we have considered the resistance corresponding to arc (see Fig. 7) as the grain-boundary resistance and the grain boundary contributions had been managed to be distinguished using the ZSimpWin software by modeling the IS plots with the equivalent circuit R b (R gb Q gb )(R ele Q ele ), where R b , R gb , R ele , Q gb , Q ele represent grain resistance, grain boundary resistance, electrode resistance, constant phase element of the grain boundary and constant phase element of the electrode, respectively. Above 500°C, only the total resistance was considered due to only small part of the grain-boundary contributions could be observed and the equivalent circuit LR t (Q ele R ele ) (L is inductance and R t = R b + R gb ) was used to fit the impedance spectrum in view of the contribution of cable. The bulk, grain boundary and total conductivity were then calculated according to the relation, σ i = l/SR i .
Figure 10 shows Ca content (x) dependence of the grain boundary conductivity at low temperatures. It can be seen that the grain boundary conductivity increased gradually at x < 0.1, and then decreased at x > 0.1. This may be explained by the fact that Ca doping apparently changed the amount of insulating grain-boundary phase. Addition of small amount of Ca could decrease the grain-boundary resistance, and therefore improved the grain boundary conductivity. An enhanced effect with small amount of CaO had also been observed in the Ca co-doped Ce0.8Y0.18Ca0.02O2-δ composition [24] and CGO20 system [25]. As we could not observe the grain-boundary contributions at temperatures above 500°C, we have mainly concentrated on the total conductivity exhibited by the studied compositions. The temperature dependence of the total conductivity was plotted in Fig. 11 based on the Arrhenius equation. It can be seen that co-doped samples showed higher conductivities than singly doped samples, which suggests that co-doping with Gd3+ and Ca2+ could lead to the enhancement of the conductivity for the studied series. Meanwhile, nearly linear Arrhenius plots over a wide temperature range indicated the presence of only one mode of oxide ion conduction in these solid electrolytes [26]. The activation energies for conduction (Ea), calculated from the line slopes in Fig. 11, were 0.601, 0.593, 0.58, 0.615 and 0.634 eV, respectively. It can be seen that the activation energies of Ca and Gd co-doped ceria are lower than that of singly doped ceria. Moreover, the Ea value decreases to a minimum at x = 0.1. The change of the activation energies on composition seemed to be correlated with the compositional variations of conductivity—the maximum values of conductivity corresponded to the minimum energy activation.

Ca content dependence of the grain boundary conductivity (σ gb) for Ce0.8Gd0.2–x Ca x O2-δ ceramics

Arrhenius plots for Ce0.8Gd0.2–x Ca x O2-δ (x = 0–0.2) pellets sintered at 1400°C
Figure 12 further depicts the isothermal total conductivity dependence of the Ca-doping content for Ce0.8Gd0.2–x Ca x O1.9-δ sintered at 1400°C. As can be seen, unlike the unique effect of calcium co-doping in Ce0.8Sm0.2–x Ca x O1.9 system reported by S. Banerjee [17], there emerged a maximum in the electrical conductivities of Ce0.8Gd0.2–x Ca x O1.9-δ , i.e. Ce0.8Gd0.1Ca0.1O2-δ exhibits the highest total conductivity. The highest conductivity was equal to 0.0742 S/cm at 700°C, which was not only higher than that of single-doped Ce0.8Gd0.2O1.9 (0.0611 S/cm) and that of single-doped Ce0.8Ca0.2O2-δ (0.0376 S/cm) but also comparable with its analogue reported by Sujatha [27].

The isothermal conductivity plots with Ca content (x) for Ce0.8Gd0.2–x Ca x O2-δ sintered at 1400°C
Although above 600°C, CeO2 always shows electronic conductivity and the conductivity at higher temperature may be mixed ionic and electronic conductivity, the results of the ionic transfer number measurements for the similar ceria-based electrolyte demonstrated that the conductivity of doped-ceria was predominantly ionic in the area of high oxygen partial pressure, as reported elsewhere [28–30]. In order to further understand the mechanism behind the enhancement of conductivity, the valance states of Ca-and Gd-codoped CeO2 were identified by X-ray photoelectron spectroscopy (XPS). Figure 13 shows the high resolution photoelectron spectra of the Gd 4d, Ca 2p and O 1 s core levels of representative sample Ce0.8Gd0.10Ca0.10O2-δ . As can be seen from Fig. 13(a), three peaks can be observed for the Gd 4d core level spectrum after deconvolution of the experimentally measured curve. Bonding energy of 145.5 eV and 140.3 eV are attributed to Gd 4d3/2 and Gd 4d5/2 while the rest peak at 152.6 eV can not be given the exact reason [31]. The Ca 2p core level spectrum with the deconvolution results is shown in Fig. 14(b). The Ca 2p3/2 and Ca 2p1/2, due to spin orbit splitting, are clearly distinguishable at around 346.6 and 350.0 eV, respectively [32]. The oxygen 1 s core level spectrum with the fitted curve is shown in Fig. 13(c). The peak at 529.6 eV, which is very close to the literature value 529.5 eV [31, 33], is ascribe to the lattice oxygen. Another deconvolution peak at 531.9 eV may be assigned to some hydroxyl like groups, which presumably result from surface bonded water [31, 34]. Figure 14 shows the Ce 3d XPS spectra of all the compositions investigated. The corresponding bonding energies along with literature data are given in Table 2. Comparing with literature results [31, 32] and with the single oxides CeO2 and Ce2O3 [35–37], six peaks (labeled as v0, v1, v2 (Ce 3d 5/2), v′0, v′1 and v′2 (Ce 3d 3/2) with vertical line) are found in samples Ce0.8Gd0.2–x Ca x O1.9-δ with x = 0, 0.05, 0.10, indicating that the surface of these samples contains virtually pure CeO2. Additional two peaks at 885.2 eV and 903.8 eV appeared in sample Ce0.8Gd0.05Ca0.15O2-δ and sample Ce0.8Ca0.2O2-δ , suggesting the coexistence of CeO2, plus Ce2O3 species. Presence of a trace of Ce3+ in the surface of Ce0.8Gd0.05Ca0.15O2-δ and Ce0.8Ca0.2O2-δ may be related to a chemical reduction due to increasing the dopant Ca concentration [32]. It should be mentioned that it is difficult to quantitatively determine the Ce3+ fraction due to the complexity of the Ce3d spectrum. Nevertheless, the reduction level of Ce4+ to Ce3+ is very low, judging qualitatively from the u1 and u′1 peak area.

High resolution XPS spectra of Gd 4d, Ca 2p and O 1 s regions for Ce0.8Gd0.1Ca0.1O2-δ

Ce 3d XPS spectra from the co-doped Ce0.8Gd0.2–x Ca x O2-δ samples: (a) x = 0, (b) x = 0.05, (c) x = 0.10, (d) x = 0.15 and (e) x = 0.20
On the basis of the above-mentioned XPS analysis, we can deduce that the existence of Gd3+, Ca2+ and Ce4+ (with a trace of Ce3+ or without of Ce3+) in the investigated surface region of the materials will form oxygen vacancies for compensating the deficiency in positive charge due to doping with gadolinium and calcium. Oxygen vacancy, but not electron, is the dominant conduction mechanism. The results of XPS are thus in accordance with that of ionic transfer number measurements. But, the conclusion drawn from the results of XPS should not be overrated since the analysis is limited to the surface. To get the exact valance of cerium, more bulk sensitive characterization techniques such as X-ray absorption spectroscopy or nuclear magnetic resonance should be employed.
In order to illuminate the variation of the total conductivity exhibited by the Ca-doping effect, the free volume (FV) available in the lattice (see Table 1) was used to try to explain the trend of the conductivity. The concept of free volume was first introduced by Sammells [38] and subsequently applied to discuss the electrical conductivity of perovskite-based solid electrolytes by Nomura [39] and Yamamura [40]. Recently, this idea was successfully applied to fluorite-type structures, such as Ce0.8Sm0.2–x Ca x O2-δ [17]. Generally, the conductivity increases with increase of FV. For instance, FV present in Ce0.8Sm0.2–x Ca x O1.9 exhibited a systematic increase with increasing Ca content and the conductivity was enhanced accordingly. However, an increase of FV in this investigated series did not give rise to consistently increase of conductivity. This may be caused by the effect of oxygen vacancies in the lattice. As evident from Table 1, partial substitution of Gd3+ with Ca2+ ion will bring more oxygen vacancies in the lattice because of the charge compensation in electrolyte materials, which could be expressed with following equations by using kröger-Vink notation:
The vacancy (\( V_O^{{ \cdot \cdot }} \)) with positive charge maybe attract the doping ions (\( Gd_{{Ce}}^{'} \)and\( Ca_{{Ce}}^{{''}} \)) with negative charge to produce clusters (\( Gd_{{Ce}}^{'} V_O^{{ \cdot \cdot }}/Gd_{{Ce}}^{'} V_O^{{ \cdot \cdot }}Gd_{{Ce}}^{'} \) and \( Ca_{{Ce}}^{{''}}V_O^{{ \cdot \cdot }}/Ca_{{Ce}}^{{''}} V_O^{{ \cdot \cdot }}Ca_{{Ce}}^{{''}} \)) due to the electrostatic attraction [4]. But with too much Ca2+ ions doping, the defect association (e.g., \( Ca_{{Ce}}^{{''}}V_O^{{ \cdot \cdot }}/Ca_{{Ce}}^{{''}}V_O^{{ \cdot \cdot }}Ca_{{Ce}}^{{''}} \)) near the Ca2+ might begin to form at the expense of oxygen vacancies [4], and the clusters might prevent oxygen vacancies from passing through the lattice and consequently the conductivity decreased.
Therefore, the partial substitution of Gd3+ ions with Ca2+ ions caused two opposite effects for the conductivity. On the one hand, the increase of Ca content could lead to the increment of FV which resulted in the increase in the conductivity. On the other hand, with more Ca ions displacement, the defect association might begin to form at the expense of oxygen vacancies. The formation of defect association may prevent oxygen vacancies from passing through the lattice, and finally caused the decline of conductivity.
4 Conclusions
Co-doped ceria of Ce0.8Gd0.2−x Ca x O2-δ (x = 0–0.2) solid solutions with fluorite structure were prepared by oxalate co-precipitate method. The nanosized powders can be obtained after being calcined at 600°C. The relative densities of the representative Ce0.8Gd0.1Ca0.1O2−δ pellets sintered at different temperatures were measured and the optimal density was above 96%. The corresponding SEM images showed relatively uniform grains with grain size from 0.5–1 μm to 1–2 μm. Impedance spectroscopy measurements indicated that in comparison to the singly doped ceria, co-doped ceria showed much higher ionic conductivities at 250–800°C. The conductivities of Ce0.8Gd0.2−x Ca x O2-δ system reached a maximum at x = 0.1. At 700°C, the ionic conductivity of Ce0.8Gd0.1Ca0.1O2-δ was 0.0742 S/cm. It indicates that co-doing with an appropriate ratio of Gd3+ and Ca2+ could effectively improve the ionic conductivity of ceria-based electrolytes.
References
R.M. Ormerod, Chem. Soc. Rev. 32, 17 (2003)
D.J.L. Brett, A. Atkinson, N.P. Brandon, S.J. Skinner, Chem. Soc. Rev. 37, 1568 (2008)
J.C. Boivin, G. Mairesse, Chem. Mater. 10, 2870 (1998)
H. Inaba, H. Tagawa, Solid State Ion. 83, 1 (1996)
V.V. Khartona, F.M.B. Marquesa, A. Atkinson, Solid State Ion. 174, 135 (2004)
J.W. Fergus, J. Power. Sources 162, 30 (2006)
M. Mogensen, N.M. Sammes, G.A. Tompsett, Solid State Ion. 129, 63 (2000)
S. Omar, E.D. Wachsman, J.C. Nino, Solid State Ion. 177, 3199 (2006)
S.K. Tadokoro, E.N.S. Muccillo, J. Eur. Ceram. Soc. 27, 4261 (2007)
X. Sha, Z. Lü, X. Huang, J. Miao, Z. Ding, X. Xin, W. Su, J. Alloy. Compd. 428, 59 (2007)
X. Guan, H. Zhou, Z. Liu, Y. Wang, J. Zhang, Mater. Res. Bull 43, 1046 (2008)
Y. Liu, B. Li, X. Wei, W. Pan, J. Am. Ceram. Soc. 91, 3926 (2008)
X. Guan, H. Zhou, Y. Wang, J. Zhang, J. Alloy. Compd. 464, 310 (2008)
W. Zając, J. Molenda, Solid State Ion. 179, 154 (2008)
Y. Zheng, H. Gu, H. Chen, L. Gao, X. Zhu, L. Guo, Mater. Res. Bull 44, 775 (2009)
H. Yamamura, E. Katoh, M. Ichikawa, K. Kakinuma, T. Mori, H. Haneda, Electrochemistry 68, 455 (2000)
S. Banerjee, P.S. Devi, D. Topwal, S. Mandal, K. Menon, Adv. Funct. Mater. 17, 2847 (2007)
S. Banerjee, P.S. Devi, Solid State Ion. 179, 661 (2008)
R.W.M. Kwok, Department of Chemistry, The Chinese University of Hong Kong, Shatin, Hong Kong, Available from: http://www.uksaf.org/software.html
R.O. Fuentes, R.T. Baker, J. Power. Sources 186, 268 (2009)
R.D. Shannon, Acta Crystallogr. A32, 751 (1976)
E. Barsoukov, J.R. Macdonald, Impedance Spectroscopy Theory, Experiment, and Applications (John Wiley & Sons, Inc, 2005), p. 246
J.T.S. Irvine, D.C. Sinclair, A.R. West, Adv. Mater. 2, 132 (1990)
A. Moure, J. Tartaj, C. Moure, J. Eur. Ceram. Soc. 29, 2559 (2009)
S. Molin, M. Gazda, P. Jasinski, J. Rare Earth. 27, 655 (2009)
W. Huang, P. Shuk, M. Greenblatt, Chem. Mater. 9, 2240 (1997)
P. Sujatha Devi, S. Banerjee, Ionics 14, 73 (2008)
S. Dikmen, P. Shuk, M. Greenblatt, Solid State Ion. 126, 89 (1999)
S. Wang, T. Kobayashi, M. Dokiya, T. Hashimotob, J. Electrochem. Soc. 147, 3606 (2000)
X. Guo, W. Sigle, J. Maier, J. Am. Ceram. Soc. 86, 77 (2003)
P. Datta, P. Majewski, F. Aldinger, Mater. Character. 60, 138 (2009)
M. Yan, T. Mori, J. Zou, F. Ye, D.R. Ou, J. Drennan, Acta Mater. 57, 722 (2009)
J.L.M. Rupp, T. Drobek, A. Rossi, L.J. Gauckler, Chem. Mater. 19, 1134 (2007)
A. Kossoy, H. Cohen, T. Bendikov, E. Wachtel, I. Lubomirsky, Solid State Ion. 194, 1 (2011)
G.M. Iogo, E. Paparazzo, O. Bagnarelli, N. Zacchetti, Surf. Interf. Anal. 16, 515 (1990)
J.E. Fallah, L. Hilaire, M. Romeo, F.L. Normand, J. Electron. Spectrosc. Relat. Phenom. 73, 89 (1995)
L. Qiu, F. Liu, L. Zhao, Y. Ma, J. Yao, App. Surf. Sci. 252, 4931 (2006)
A.F. Sammells, R.L. Cook, J.H. White, J.J. Osborne, R.C. MacDuff, Solid State Ion. 52, 111 (1992)
K. Nomura, S. Tanase, Solid State Ion. 98, 229 (1997)
H. Yamamura, K. Matsui, K. Kakinuma, T. Mori, Solid State Ion. 123, 279 (1999)
Acknowledgements
We would like to thank the financial supports of the National Natural Science Found of China (Nos. 20971111, 21001096 and J0830412).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhao, XL., Liu, JJ., Xiao, T. et al. Effect of Ca co-dopant on the electrical conductivity of Gd-doped ceria. J Electroceram 28, 149–157 (2012). https://doi.org/10.1007/s10832-012-9696-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10832-012-9696-5