
Abstract
Increasing concentrations of the anaesthetic agent propofol initially induces sedation before achieving full general anaesthesia. During this state of anaesthesia, the observed specific changes in electroencephalographic (EEG) rhythms comprise increased activity in the δ− (0.5−4 Hz) and α− (8−13 Hz) frequency bands over the frontal region, but increased δ− and decreased α−activity over the occipital region. It is known that the cortex, the thalamus, and the thalamo-cortical feedback loop contribute to some degree to the propofol-induced changes in the EEG power spectrum. However the precise role of each structure to the dynamics of the EEG is unknown. In this paper we apply a thalamo-cortical neuronal population model to reproduce the power spectrum changes in EEG during propofol-induced anaesthesia sedation. The model reproduces the power spectrum features observed experimentally both in frontal and occipital electrodes. Moreover, a detailed analysis of the model indicates the importance of multiple resting states in brain activity. The work suggests that the α−activity originates from the cortico-thalamic relay interaction, whereas the emergence of δ−activity results from the full cortico-reticular-relay-cortical feedback loop with a prominent enforced thalamic reticular-relay interaction. This model suggests an important role for synaptic GABAergic receptors at relay neurons and, more generally, for the thalamus in the generation of both the δ− and the α− EEG patterns that are seen during propofol anaesthesia sedation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
General anaesthesia (GA) is a reversible medical procedure that is commonly induced by the administration of a combination of anaesthetic agents (AAs) to induce amnesia, sedation, hypnosis (loss of consciousness) and immobility in patients. There are differing definitions of general anaesthesia, but phenomena such as analgesia, muscle relaxation and anxiolysis should be included, in addition to the most obvious effect of loss of consciousness (Rudolph and Antkowiak 2004). Although GA is used for surgery, its precise underlying mechanisms are not yet fully understood.
Propofol, an emulsion formulation of 2,6-di-isopropylphenol, is a short-acting, intravenously administered hypnotic agent; and is in widespread use for sedation and general anaesthesia because of its relatively fast onset and offset, and anti-emetic effects (San-Juan et al. 2010). Clinically relevant concentrations of propofol cause specific changes in electroencephalogram (EEG) rhythms of healthy adult humans. Up to sedative concentrations, these observations include an increase in power in the α− (at about 10 Hz) and δ− (at about 3 Hz) frequency bands over the frontal head region, accompanied by decreased α−activity, and an increase in δ−activity over the occipital sites (Murphy et al.2011; Ching et al.2010; Feshchenko et al. 2004; Hazeaux et al. 1987; Sellers et al.2013). It has been shown that in deeper sedation propofol attenuates posterior α− power and increases frontal β and total power (Cimenser et al. 2011; Gugino et al. 2001) and is associated with a frontal increase in α−, δ−, and 𝜃−power. With the loss of consciousness frontal power further increases across all frequency bands (Gugino et al. 2001).
Although it is not precisely clear yet how the specific changes in EEG rhythms occur, there have been some efforts to explain the underlying mechanims leading to such EEG-level phenomena. For instance, Alkire et al. (2000) proposed that thalamo-cortical cells are involved in producing anaesthetic induced δ−activity. They propose that decreased excitation causes the firing mode of thalamic relay neurons to shift from a tonic to a burst pattern, thereby producing δ−activity. Ching et al. (2010) have also developed a thalamo-cortical model that suggests the importance of the thalamus in rhythmic activity in the frontal EEG. They have shown that synchronous frontal α−activity in the EEG is the result of recruitment of some subset of the thalamic network into the cortical rhythm. This is in contrast to the theory of thalamic deactivation during general anaesthesia. A recent study (Vijayan et al. 2013) suggests that reduction in hyperpolarization-activated current (I h ) silences bursting of a subset of thalamo-cortical cells leading to suppression of occipital α−activity. Their work indicated that increased GABAergic inhibition onto thalamo-cortical cells resulted in an α− resonance that is reinforced by reciprocal corticothalamic feedback, and hence leads to the emergence of frontal α−activity. This frontal enhancement and occipital attenuation of α−activity is commonly termed anteriorization.
Other theoretical studies have pointed out the importance of anaesthetic-induced cortical inhibition considering (Hashemi et al. 2014; Hindriks and van Putten 2012) or neglecting (Liley and Bojak 2005; Bojak and Liley 2005; Steyn-Ross et al. 1999; Steyn-Ross et al. 2001a, 2001b; Molaee-Ardekani et al. 2007; Hutt and Longtin 2009) the feedback loop to the thalamus.
In recent decades there have been many studies on the molecular and cellular actions of anaesthetics on single receptors (Franks and Lieb 1994; Alkire et al. 2008). However, to understand how the emergent behaviours at the EEG-level (macroscopic scale) can be caused by the actions of anaesthetics at single neuron level (microscopic scale), it is necessary to establish a bridge between the two scales. A well-tried method to link these two levels of brain dynamics is using the mean-field approach to characterize the activity of large populations of neurons at an intermediate mesoscopic scale (Deco et al. 2008; Drover et al. 2010; Bressloff 2012). The EEG represents the neural activity of thousands or millions of spatially oriented pyramidal neurons of the cortex (Nunez and Srinivasan 2006). By virtue of this large number of neurons, it is reasonable physiologically to model EEG by considering spatiotemporal neural activity of populations using a mean-field approximation (Nunez 1981; Jirsa and Haken 1996; Liley et al. 2002; David and Friston 2003; Robinson et al. 1997; Robinson et al. 2001a; Robinson et al. 2002; Freestone et al. 2011; Pinotsis et al. 2012). This description involves averages of synaptic and neuron activity over a population in small spatial patches and short time windows (Nunez 1974; Wilson and Cowan 1972).
A large number of previous theoretical studies on anaesthetic effects apply a mean-field model to explain various features in EEG data recorded during general anaesthesia (Liley and Bojak 2005; Bojak and Liley 2005; Steyn-Ross et al. 1999; Steyn-Ross et al. 2001a, 2001b; Molaee-Ardekani et al. 2007; Foster et al. 2008; Hindriks and van Putten 2012; Hutt and Longtin 2009; Hashemi et al. 2014). These theoretical studies are based on various extensions of the model of Liley and Wright (1994), Wright and Liley (1996), Wilson and Cowan (1972), and Amari (1977) including excitatory and inhibitory neurons and synapses. The present work considers a neural population model based on a recently developed neural field model of anaesthetic action (Hutt and Longtin 2009). It includes excitatory and inhibitory cortical populations which are synaptically connected to thalamic reticular and relay neuron populations by delayed thalamo-cortical axonal fibres. The model takes into account anaesthetic action at inhibitory GABAergic synaptic receptors in both cortical inhibitory neurons and thalamic relay cells. A very recent model (Hashemi et al. 2014) takes into account anaesthetic action of extra-synaptic GABAergic receptors, which are however neglected here.
In order to implement the anaesthetic action in cortical and thalamic structures, a careful look at neural receptors and their response to anaesthetic action is mandatory. Recent studies have revealed possible molecular sites and neuronal pathways for the action of the anaesthetic propofol in the human brain. In vivo extracellular recordings have demonstrated that propofol suppresses field potentials in the rat thalamus and cortex (Antkowiak 2002). Although some reports put emphasis on more prominent effects in the cortex, the cortical suppression may, in fact, be secondary to anaesthetic action on projection neurons located elsewhere in the brain, especially in the thalamus. Also several lines of evidence indicate that thalamus and thalamic neuronal circuits might be important target-sites for the hypnotic effects of propofol (Franks 2008; Ying and Goldstein 2001; Zhang et al. 2002). Moreover, experimental observations strongly suggest that reticular thalamic neurons are a major source of inhibition to the relay neurons; and are thus a critical structure for the effect of propofol in thalamic circuits (Byoung-Kyong 2010; Ying and Goldstein 2005b). These findings motivated us to more concentrate on the effect of propofol in thalamic neurons - in contrast to previous studies, e.g., Hindriks and van Putten (2012), Steyn-Ross et al. (2004), and Bojak and Liley (2005). Based on these experimental observations, we first study a full model including anaesthetic effects in cortical and thalamic populations at light sedation. Then, in order to form a hypothesis that the GABAergic inhibition in thalamic cells seems to play a crucial role in the generation of characteristic EEG patterns under propofol sedation, we neglect the anaesthetic action in the cortex in a reduced model. This assumption points out the importance of the thalamus for major neural effects under anaesthesia sedation as well as simplifies the model under study, while later results shall reveal that the model is still adequate to reproduce the observed changes in EEG rhythms. The model aims to reproduce spectral features in light sedation, i.e. at low propofol concentrations. This anaesthetic phase does not exhibit increased spectral power in the β frequency band, that is sometimes called beta buzz.
The study provides evidence that thalamic GABAergic synaptic inhibition is one of the key actions inducing the characteristic changes in the δ− and α−frequency ranges under propofol-induced sedation. Moreover, the detailed study of sub-circuits suggests the existence of two major cortico-thalamic sub-circuits that generate δ− and α−spectral power peaks. To our best knowledge, this is the first neural population study that reproduces the EEG changes over frontal and occipital regions in the thalamo-cortical system.
The following section presents the acquisition procedure of experimental EEG under anaesthesia and the neural population model involving the population network, the anaesthetic action and the analysis steps to be taken. Then, we present the power spectra of experimental EEG in frontal and occipital electrodes and show that both the proposed full and reduced neural population models reproduce very well the characteristic spectral features in the δ− and α−band subjected to the propofol concentration. The subsequent analysis of the model reveals the importance of multiple resting states and the presence of effective sub-circuits generating the spectral features in δ− and α−frequency bands.
2 Materials and methods
2.1 EEG recordings during anaesthesia sedation
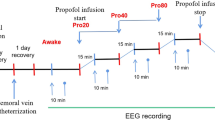
We re-analysed previously-obtained experimental data from subjects that had been given a short propofol anaesthetic. The details of the methods can be found in Johnson et al. (2003). After obtaining regional ethical committee approval and written informed consent, five healthy subjects (mean age 27.7 yrs, four males) were included in the study. They were on no psychoactive drugs and had been starved for at least 6 hours prior to the study, and were monitored and managed as per the Australia and New Zealand College of Anaesthetists best practice guidelines. The induction consisted of an intravenous infusion of propofol at 1500 mg/hr until the subject no longer responded to verbal command (usually after about 5 minutes). At this point the propofol infusion was stopped and the subject allowed to recover spontaneously (typically after about 5 minutes). The estimated effect-site concentrations of propofol were calculated using standard population-based pharmacokinetic models.
High-density EEG was acquired using the Electrical Geodesics 128 channel Ag/AgCl electrode system (Eugene, CO, USA) referenced to Cz. Electrode impedances were below 30 KOhm (100 MOhm input impedance amplifier), and a custom built head support device was used so as to allow recording of occipital channels when supine. The sampling frequency was 250 Hz, with a 0.1−100 Hz analogue band pass filter, and 12 bit A-D conversion. The EEG data were re-referenced to a grand mean, and band-pass filtered using 3-rd order Butterworth filters 0.2−45 Hz to eliminate line-noise. A Whittaker filter was applied to reduce movement and blink artifacts. The power in each frequency was expressed in decibels, and obtained applying a short-time Fourier transform using the “spectrogram.m” Matlab (Mathworks, Natick, Massachusetts, USA) function with a moving window of 2 sec and 1 sec overlap.
2.2 Thalamo-cortical model
The body of the work is based on a thalamo-cortical neural population model that is illustrated in Fig. 1. The model consists of four different populations of neurons (Rennie et al. 2002; Robinson et al. 2001a; Robinson et al. 2002; Victor et al. 2011): cortical pyramidal neurons (E), cortical inhibitory neurons (I), thalamo-cortical relay neurons (S), and thalamic reticular neurons (R). The thalamo-cortical relay neurons receive input from, and project onto the cortical pyramidal neurons. They also project onto the thalamic reticular neurons. The cortical inhibitory neurons inhibit pyramidal neurons and receive excitatory input from them. The thalamic reticular neurons inhibit the thalamo-cortical relay neurons, and receive excitatory input from the pyramidal neurons. The excitatory connections between cortex and thalamus are associated with a conduction delay while no delay is present in the projections within the thalamus and within the cortex (Victor et al. 2011).

Schematic of the thalamo-cortical model. The blue arrows indicate excitatory connections and the red connections with filled circle ends represent inhibitory connections. The model consists of four types of neural populations, namely, cortical excitatory and inhibitory neurons, thalamo-cortical relay, and thalamic reticular neurons denoted by E, I, S, and R, respectively. In addition, the connections between cortex and thalamus are associated with the same nonzero time delay τ
The underlying model considers an ensemble of neurons on a mesoscopic scale which includes two types of neurons, namely excitatory and inhibitory cells (Hutt and Longtin 2009). The connection elements between neurons are excitatory and inhibitory chemical synapses in which both types of synapses may occur on dendritic branches of both cell types. The present model considers AMPA/NMDA and GABA A synaptic receptors and assumes spatially extended populations of both neuron types.
Mean excitatory and inhibitory postsynaptic potentials V e(x,t) and V i(x,t) at spatial location x and time t are evoked by incoming spike trains and obey
where P e (x,t) and P i (x,t) represent the mean presynaptic population firing rates at position x and time t. The functions h e (t) and h i (t) indicate the synaptic response function of excitatory and inhibitory synapses, respectively,
where 1/α e and 1/α i are the characteristic rise times of the response function for excitatory and inhibitory synapses, respectively, and 1/β e and 1/β i are the corresponding characteristic decay times. The function f j (p) reflects the action of propofol on the GABAergic synaptic receptors and is proportional to the charge transfer in these receptors. From Fig. 1, afferents from thalamic (T) reticular neurons and cortical (C) inhibitory neurons terminate at GABAergic synaptic receptors, i.e., j∈{C,T}. The function f j will be estimated from experimental data in the following subsection. The parameters a e and a i denote the synaptic gain in the absence of anaesthetics.
Following Robinson et al. (2001a), Robinson et al. (2002), and Hutt et al. (2003), the integral equations (1) may be formulated as differential equations
with
External input to the system originates from other neural populations and is considered as a non-specific input to relay neurons as
where I 0 indicates its mean value and ξ(x,t) is Gaussian uncorrelated noise with
where 〈.〉 denotes the ensemble average and κ is the intensity of the driving noise.
Moreover, the mean population firing rates P e,i in Eq. (1) depend on the effective mean potential in the corresponding populations. The present study considers a recently derived transfer function of a population of type I-neurons (Hutt and Buhry 2014)
with
where S max is the maximum population firing rate, V th is the mean firing threshold of neurons and σ is related to the standard deviation of firing thresholds. The parameter ρ<∞ reflects the properties of type I-neurons where ρ→∞ yields the standard formulation assuming McCulloch-Pitts neurons (Hutt 2012) with S i g j (V,ρ→∞)=0. The transfer function (7) takes into account the distribution of firing thresholds of the neurons in the population (Wilson and Cowan 1972) and the more similar the firing thresholds in the neural populations the smaller σ and the steeper the sigmoidal function. In contrast to the standard sigmoid function, the transfer function (7) is not anti-symmetric to its inflection point anymore (Hutt 2012) and exhibits a larger nonlinear gain (slope) for large potentials V>V th compared to small potentials V<V th, as illustrated in Fig. 2. Typically the sigmoidal shape of the transfer function leads to multiple resting states and its asymmetry gives the resting states that are closer to the V th. This asymmetry results from the firing properties of type-I neurons, see Hutt and Buhry (2014) and Hutt (2012) for more details. In this context, we mention another previous transfer function proposed by Freeman (1979) to take into account an average subthreshold value of sodium channels. We identify the mean population firing rates P e,i with the transfer function S j .

The novel population firing rate S(V) taken from Eq. (7) and the corresponding nonlinear gain d S(V)/d V in comparison to the well-known sigmoid function S i g(V,0). Panel a illustrates the mean firing rate functions and panel b presents the corresponding nonlinear gains for the standard population firing rate (blue lines) and the novel transfer function (green lines). Parameters are S max=100 Hz, V th=15 mV, σ=10 mV, ρ=0.05 mV −1
2.3 Effect of propofol on neural populations
According to experimental results, it is now widely accepted that anaesthetic agents act by binding directly to specific protein targets (Chau 2010). While it has been reported that many receptors and molecular targets contribute to general anaesthesia, there is a general agreement that GABA A receptors are important in mediating the inhibition during propofol administration (Zhou et al. 2012). It has been shown that propofol increases the decay time constant of GABA A synapses, and hence increases the total charge transfer in these synapses (Franks 2008). Interestingly, Kitamura et al. (2002) have shown experimentally that propofol has a negligible effect on the amplitude of synaptic response functions in cortical neurons, whereas it markedly increases amplitude, decay time, and thus charge transfer of GABA A receptors of inhibitory synapses located in relay neurons in the thalamic ventrobasal complex (Ying and Goldstein 2005a). Since GABA A receptors mediate most of the inhibitory synaptic transmission, the present model assumes that the inhibitory synaptic transmission within cortex and thalamus are mediated by GABA A receptors and GABA B receptors are neglected.
In order to mimic the latter experimental findings, we assume that increasing propofol concentration decreases the decay rate of the inhibitory synaptic response function β i by \(\beta _{i}= {{\beta _{i}^{0}}}/{p}\) with p≥1 (Steyn-Ross et al. 1999), where \({\beta _{i}^{0}}\) denotes the inhibitory decay rate in the absence of propofol. The factor p reflects the on-site concentration of propofol in the neural populations and p=1 indicates the baseline condition. Increasing p leads to a decrease in the decay rate constant of inhibitory synapses and thereby an increase in the charge transfer in these synapses.
Cortical inhibitory synapses retain their response amplitude while changing the anaesthetics concentration (Kitamura et al. 2002; Hutt and Longtin 2009) leading to the relative increase of the charge transfer
with
This choice fixes the maximum of cortical response function to \(h_{C}^{max}=a_{i} {\Gamma }(\alpha ,{\beta ^{0}_{i}})\). Conversely propofol increases the amplitude of evoked inhibitory post-synaptic currents in the ventrobasal relay neurons (Ying and Goldstein 2005b). To take this effect into account the relative increase of the charge transfer in thalamic G A B A A receptors is
with the relative amplitude A r (p). This choice determines the maximum of thalamic response function to \(h_{T}^{max}=a_{i} {\Gamma }(\alpha ,{\beta ^{0}_{i}}) A_{r}(p)\), i.e., the maximum response increases with A r (p) under the condition A r (1)=1. This choice is motivated by previous studies of cortical GABAergic synapses (Hashemi et al. 2014; Hindriks and van Putten 2012). An optimal fit of A r to experimental data yields A r (p)≈1.55ln(1.49+0.42exp(p))≈p 0.42, cf. Fig. 3. The response functions h i (t) for cortical and thalamic receptors subject to the anaesthetic concentration factor p are displayed in Fig. 4 illustrating the increasing response amplitude for thalamic receptors compared to the cortical receptors.

Effect of propofol on thalamic GABA A -receptors extracted from a previous experimental study (Ying and Goldstein 2005b, Fig. 5). a The factor \(p={\beta ^{0}_{i}}/\beta _{i}\) of evoked IPSCs in a relay neuron (taken from the experimentally measured normalized decay time) subject to the propofol on-site concentration c (red square) and the corresponding fitted function (blue line) \(p(c)=k_{1}\ln (k_{2}+k_{3}c)\). b The experimentally measured normalized amplitude A(c)/A 0 of evoked IPSCs subject to the propofol on-site concentration c (red square) and the corresponding fitted function (blue line) \(A(c)/A_{0}=k_{4}\ln (k_{5}+k_{6}c)\). Here A 0=A(c=0). c Since the constants k 1,…,k 6 are fitted, \(A_{r}(p)=A(p)/A_{0}=k_{4}\ln (k_{7}+k_{8}\exp (p/k_{1}))\) (red line) with k 7=k 5−(k 2 k 6/k 3), and k 8=k 6/k 3. An additional fit reveals A(p)/A 0≈p 0.42 (blue line)

The temporal synaptic response function of inhibitory GABA A synapses subject to the factor p. The factor p reflects the anaesthetic propofol concentration. a The decay phase of the cortical response function h i (t) is prolonged at larger values of p, whereas the maximum height of h i (t) with Eq. (9) is constant. b The amplitude of h i (t) in thalamic receptors with Eq. (10), i.e. A r (p)≈p 0.42, increases as a function of p. Parameters are α i =400 Hz and β i =40 Hz
We point out that, by definition, the charge transfer in the respective receptors is proportional to f C (p) and f T (p). Since the charge transfer reflects the level of inhibition, the relation of thalamic to cortical inhibition
is an increasing function of p, i.e. the larger the anaesthetic concentration the stronger is the anaesthetic-induced thalamic inhibition compared to cortical inhibition.
Finally, we mention that the propofol concentrations in Fig. 3 are equivalent to the interval [0;0.53μg/ml] for a propofol molar mass of 178g. This concentration is the on-site concentration determined in the in-vitro experiment of Ying and Goldstein (2005b). This concentration is not necessarily similar to the blood-plasma propofol concentrations given later in Fig. 5 due to pharmaco-dynamic effects, e.g. caused by the blood-brain barrier. The uncertain relationship between on-site concentrations and blood plasma concentrations represents an uncertainty in the current model.

Physiological and electroencephalographic data observed in a single subject while increasing the propofol concentration. a Blood plasma concentration of propofol with respect to administration time. Since average clinical propofol concentrations in sedation are around or smaller than 1μg/ml (Reed et al. 1996), the subject my leave the clinical sedation phase at about t=5min. b Mean spectral power in the δ− and α−frequency bands in single electrodes located along the scalp mid-line with respect to administration time. c Spectrogram of power averaged over four frontal (left) and four occipital (right) electrodes. The vertical white lines denote 2 sec-time windows at t=2 min 6 sec and at t=5 min. d Average power spectra over frontal (left) and occipital (right) electrodes during the baseline state (awake) and during sedation. The peak at about 2 Hz has been verified by modifying the sliding window duration of the power spectral density estimation technique. e Spectral power amplitude averaged over the δ− and α−frequency bands in the frontal and occipital scalp. Grey and black color encodes awake and sedation, respectively
2.4 Model equations
The mean potentials \({V_{a}^{c}}\), for a∈{E,I,R,S} in the cortical pyramidal neurons (E), cortical inhibitory neurons (I), the thalamo-cortical relay neurons (S) and thalamic reticular neurons (R) obey
evoked at excitatory (c=e) and inhibitory (c=i) synapses with \(K_{ab}(x)*S_{j}[V(x,t)]={\int }_{\Omega } K_{ab}(x-y) S_{j}[V(y,t)]dy\). The model does not distinguish inhibitory synapses on excitatory and inhibitory neurons, for simplicity, which however is possible to implement easily by increasing the number of model equations. The spatial kernel functions K a b (x−y) reflect the synaptic connection strengths in population a originating from population b in the spatial domain Ω. According to previous studies we assume that the EEG can be described in a good approximation by spatially constant neural population activity (Robinson et al. 2001a; Robinson et al. 2002), we choose K a b (x−y)=K a b δ(x−y) with the Dirac function δ(⋅) and thereby K a b (x)∗S[V(x,t)]=K a b S[V(x,t)]. In addition, the parameter τ denotes the transmission time delay between cortex and thalamus. The mean firing rate function (7) in the cortex S C [⋅] is different to the thalamic firing rate function S T [⋅], while it is identical in the cortex and for relay and the reticular populations. The nominal values for two parameter sets are displayed in Table 1 in the absence of anaesthetics.
2.5 Theoretical power spectrum
Under the assumption of the spatial homogeneity, mean post-synaptic potentials in Eqs. (11) do not depend on spatial locations and then obey the general delay differential equation system
in which \(\boldsymbol {X}(t)=({V^{e}_{E}},{V^{i}_{E}},{V^{e}_{I}}, {V^{i}_{I}},{V^{e}_{S}}, {V^{i}_{S}}, {V^{e}_{R}})^{\top }\in \mathbb {R}^{N}\) is the activity variable vector with dimension N=7. The high index ⊤ denotes the transposed vector or matrix. The nonlinear vector function \(\mathbf {f}\in \mathbb {R}^{N}\) includes the nonlinear transfer functions and depend on the anaesthetic factor p. The external input is written as \(\boldsymbol {I} (t)=\boldsymbol {I}_{0}+\boldsymbol {\xi }(t)\in \mathbb {R}^{N}\) with I 0=(0,0,0,0,I 0,0,0)⊤ and ξ(t)=(0,0,0,0,ξ(t),0,0)⊤. The diagonal matrix operator \(\hat {\boldsymbol {L}}(\partial /\partial t,p) \in \mathbb {R}^{N \times N}\) includes all temporal operators \(\hat {L}_{e,i}\) and depends on the anaesthetic factor p.
Assuming small random fluctuations ξ(t), we define the resting state \(\boldsymbol {X}_{0} \in \mathbb {R}^{N}\) as a fixed point of Eq. (12), for which \(\hat {\boldsymbol {L}}=\boldsymbol {1}\), and which is given by
We observe that the resting state depends on p and hence on the anaesthetic concentration (see Appendix A). The current model follows the standard assumption (Nunez and Srinivasan 2006; Robinson et al. 2001a; Robinson et al. 2002; David and Friston 2003; David et al. 2006) that the electric dipol generating the EEG is modelled well by fluctuations of the dendritic currents about the resting state. Hence small deviations \(\boldsymbol {Y}(t) \in \mathbb {R}^{N}\) from the resting state X 0 generate the EEG and obey
where A(p), \(\boldsymbol {B}(p) \in \mathbb {R}^{N \times N}\) are constant matrices. Following previous studies (Nunez and Srinivasan 2006; Robinson et al. 2001a; Robinson et al. 2002; Robinson et al. 2004; Rennie et al. 2002; David and Friston 2003), the EEG is generated by the population activity of pyramidal cortical cells \({V^{e}_{E}}\). Then, by virtue of the specific choice of external input to relay neurons, the power spectrum of the EEG at certain frequency ν reads (Hutt 2013)
where P E just depends on one matrix component of \(\boldsymbol {\tilde {G}}(\nu ,p)\), see Appendix B for its definition. The matrix element \({\tilde {G}}_{1,5}(\nu ,p)\) depends primarily on the level of excitation and inhibition in the various populations and on the nonlinear gains d S T,C [V]/d V computed at the resting state of the system. Since the resting state depends strongly on the anaesthetic concentration factor p, in turn the nonlinear gains computed at the resting state depends on p as well.
2.6 Frequency response function
The EEG results from the interactions of various neural populations which may be viewed as interacting sub-circuits (Robinson et al. 2001a; Robinson et al. 2002; Hindriks and van Putten 2013). In the full model (Fig. 1), we identity the sub-circuits E→S→E (cortico-thalamic relay circuit), E→I→E (intra-cortical circuit), S→R→S (intra-thalamic circuit) and E→R→S→E (cortico-thalamic circuit). To reveal the relative contribution of these sub-circuits to the spectral power in the excitatory cortical population in certain frequency bands, we define the relative frequency response function from neurons of type b to neurons of type a
where l∈{e s e,e i e,s r s,e s r e}, \(\eta _{ese}=\chi ^{e}_{es}\chi ^{e}_{se}\), \(\eta _{eie}=\chi ^{i}_{ei}\chi ^{e}_{ie}\), \(\eta _{srs}=\chi ^{i}_{sr}\chi ^{e}_{rs}\), and \(\eta _{esre}=\chi ^{e}_{es}\chi ^{i}_{sr}\chi ^{e}_{re}\), and
The relative frequency response function ψ l defined in Eq. (16) is motivated by a similar measure defined by Hindriks and van Putten (2012) but extends this previous definition by its frequency dependence. Here \(S_{C,T}^{\prime }(V^{\ast e,i}_{b})\) are the nonlinear gains, i.e. the derivative of the mean firing rate function with respect to the voltages \(V^{e,i}_{b}\) computed at the resting states \(V^{\ast e,i}_{b}\). The term e −2πiντ denotes the phase shift due to the propagation delay between cortex and thalamus. Since it does not reflect any changes in our results, it can be neglected in the analysis. The functions ϕ e,i are the Fourier transform of h e,i (t) and represent the synaptic frequency responses
Increasing the factor p decreases the decay rate of synaptic inhibition β i and increases the charge transfer f j (p). Hence the enhancement of inhibition leads to increased inhibitory synaptic frequency response function \(\phi ^{i}_{ab}\) given by Eq. (17). Moreover, the administration of propofol changes the resting states and consequently the nonlinear gain functions \(S_{C,T}^{\prime }(V^{\ast e,i}_{b})\) which may increase or decrease.
The δ− and α−frequency bands are defined in the range (0.5−4 Hz) and (8−13 Hz), respectively.
2.7 Role of different populations and anatomical loops
In the last part of the study, we consider a reduced model neglecting cortical inhibition in order to elucidate the role of thalamic inhibition. To understand the role of different populations and the anatomical circuits of the reduced thalamo-cortical model, we study the contribution of different populations to the power spectra over different brain areas. It is assumed that the system fluctuations about the resting state are a linear superposition of damped noisy oscillations whose frequencies are determined by the imaginary part of the characteristic roots of Eq. (14). To quantify the contribution of each characteristic root to the power spectrum, we select a certain root and consider the fluctuations Y(t)=[y 1(t),y 2(t),y 3(t),y 4(t)]⊤ with \(y_{n}(t)=\hat u_{n} e^{\lambda t}+ \hat {u}_{n}^{\ast } e^{\lambda ^{\ast } t},~n=1,\dots ,4\). Here λ is the root and \(\hat u_{n}\) is the n−th element of its eigenvector of the corresponding eigenvalue problem. Then for each root
reflects the power in a certain oscillation mode with root λ in population n. Since roots are associated to certain frequency bands, w n reflects the contribution of population n to the power spectrum in a certain frequency band. For instance, w 1 is the contribution of excitatory currents in cortical neurons, w 2 reflects the contribution of the excitatory currents in the relay neurons, w 3 the contribution of inhibitory currents in relay neurons and w 4 the contribution of excitatory currents in reticular neurons (see Appendix C).
3 Results
3.1 Experimental power spectrum
An example of the time course of the progressive increase in propofol effect-site concentrations, and the changes in δ− and α−power in each of the 11 para-midline electrodes are shown in Fig. 5a and b, respectively. Figure 5c shows the spectrogram of experimental EEG power averaged over frontal and occipital electrodes. The power spectra of frontal and occipital average EEG during the baseline phase and during sedation are shown in Fig. 5d. They show typical changes in EEG patterns; namely that the propofol resulted in a loss of the α−rhythm (∼10 Hz) in the occipital electrodes, but a gain in α− and β−power (peak frequency around 13 Hz) in the frontal leads (p<0.05, t-test). There is a clear line of spatial delineation between the two modes of EEG response; most of the central and parietal electrodes are similar to the occipital pattern of response. Propofol also causes an increase in δ−power in both frontal and occipital regions (p<0.05, t-test). Figure 5e summarises these results.
Figure 5d and e consider propofol concentrations which lead to sedation. In some patients, there is an increased activity in the frontal β−band which is seen at about 6 min in Fig. 5c. The alternations in higher frequencies are not specifically investigated in our model.
3.2 Power spectrum of full model
This section presents the results of the analytical study of the full model as seen in Fig. 1.
3.2.1 Frontal spectrum
Figure 6 shows the EEG power spectrum (15) in the baseline condition (absence of propofol) and after the administration of propofol for certain parameters (parameter set I in Table 1). We observe an increase of power in the δ− and α−frequency bands while increasing the propofol concentration accompanied by α−activity shifted to higher frequencies (cf. Fig. 6a). These amplitude power changes bear a strong resemblance to the empirical observations in EEG power spectrum over the frontal region (cf. Fig. 6b). These results are confirmed by numerical simulations of the model.

The theoretical and experimental EEG-spectral power in the baseline and the sedation conditions. a The solid lines indicate the analytical solutions and the dashed lines show the numerical solutions of the model system for the control condition (p=1) and under sedation (p=1.165). b The normalized power amplitude of experimental data in frontal electrodes and the model over δ− and α−band in the baseline condition (blue bars) and in the sedation condition (red bars). Parameters are taken from set I in Table 1, the spectrum is computed at the upper resting state of the system
To reveal the origin of the spectral EEG-changes with increasing concentration, we relate the maxima of spectral power to the roots of the characteristic equation of Eq. (14) λ=γ+2π i ν. Figure 7 illustrates the damping rate γ and frequency ν of the roots in the δ− and α−frequency ranges. While the power peak in α−frequency range increases by the administration of propofol, the δ−frequency is maintained for different propofol concentrations. Moreover, the damping rate of δ− and α−activity increase with increasing anaesthetic level and hence the power in the corresponding frequency band increase in accordance to standard spectral theory.

Modulation of δ− and α−model activity over the frontal region. The panels show the frequency ν in the roots imaginary part which lie in the δ−and α−frequency ranges in panels a and c, respectively. Panels b and d show the corresponding damping rates as a function of the anaesthetic factor p. The factor p reflects the anaesthetic propofol concentration
3.2.2 Occipital spectrum
For different parameters (set II in Table 1), Fig. 8 shows the model power spectrum resembling experimental activity observed in occipital EEG-electrodes. The power in the δ− frequency range increases and the α−power decreases while increasing the anaesthetic level (cf. Fig. 8b). Figure 9 presents the effect of increasing propofol concentration on damping rate and frequency of the model EEG in the δ− and α−frequency ranges. It can be observed that as propofol concentration increases, the damping rate of δ−activity increases whereas α−damping rate decreases. This finding is consistent with enhanced δ− and attenuated α−power observed experimentally. In addition, the frequency of the activity in both frequency bands increase very slightly.

The theoretical EEG-spectral power compared to the power observed experimentally in occipital EEG-electrodes in baseline and under sedation. a The solid and dashed lines indicate the analytical and numerical solutions, respectively, in the baseline (p=1) and in the anaesthesia sedation condition (p=1.06). b The blue and red bars show the normalized power amplitude of experimental data in occipital electrodes and the model over δ− and α−band in the baseline condition (blue bars) and in the sedation condition (red bars). Parameters are taken from set II in Table 1, the spectrum is computed at the lower resting state of the system

Modulation of δ− and α−activity in the model over occipital region. Shown are the frequency of δ−and α−oscillation in panels (a) and (c), respectively, and the corresponding damping rates in (b) and (d), respectively as a function of the factor p. The factor p reflects the anaesthetic propofol concentration
3.2.3 Resting state and gain function
Figure 10 presents the mean potential of pyramidal neurons in the resting state denoted by \(V^{\ast e}_{E}\), and the corresponding nonlinear gain dependent on the propofol concentration for the two parameter sets considered in the previous section. We observe that three resting states may occur in the baseline condition, cf. Fig. 10a and b. Moreover, the upper and the center resting states collide at a critical value of p. Above this critical level, there exists a single resting state only at a low mean potential. The stability study of the resting states reveal that the upper and lower resting states are linearly stable, whereas the center resting state is unstable. This shows that the system may evolve either about the upper or the lower state, but never about the center resting state.

The resting states of the system determined by Eq. (13) subject to the anaesthetic level. The panels a and b show values of \(V^{\ast e}_{E}\) in the resting states for the two parameter sets I and II, respectively. Panels c and d present the corresponding nonlinear gain function d S C /d V. The upper and lower stable branches are displayed respectively in solid blue and green lines whereas the middle unstable branches are encoded in dashed red lines. For illustration reasons, the lower branches are shown again in the insets. The factor p reflects the anaesthetic propofol concentration
Since the spectral power depends heavily on the resting state and the nonlinear gain, the distinction between the upper and lower resting states is important to understand the power changes shown in the previous section and in experiments. Increasing the anaesthetic concentration decreases the mean potential of the upper resting state and, since the upper mean potential values are larger than the mean firing threshold and by virtue of the sigmoidal shape of the transfer function, decreasing \(V^{\ast e}_{E}\) on the upper resting state leads to an increase of the corresponding nonlinear gain function (Fig. 10c and d). Conversely increasing the anaesthetic level on the lower resting state decreases the nonlinear gain.
3.2.4 Sub-circuits
Figure 11 illustrates the contribution of different anatomical loops l∈{e s e,e i e,s r s,e s r e} to the power spectrum at the upper and lower resting states of the system corresponding to the frontal and occipital regions, respectively.

The change of the relative frequency response function ψ l (ν,p) defined by Eq. (16) in the δ− and α−frequency bands within different anatomical loops of the thalamo-cortical system. The panels in a consider fluctuations about the upper resting state describing frontal EEG and panels in b give the results at the lower resting state describing occipital EEG. The factor p reflects the anaesthetic propofol concentration
In the upper resting state, increasing the propofol concentration increases the inhibitory synaptic frequency response function \(\phi ^{i}_{ab}\) (cf. Eq. (17)) as well as the nonlinear gain functions \(S_{C,T}^{\prime }(V^{\ast e,i}_{b})\) (cf. Fig. 10c). Hence, as observed in Fig. 11a, the relative frequency response function ψ l in the δ− and α−band increases for all loops. This affirms the enhancement of the power in δ− and α−band observed over frontal electrodes.
At the lower resting state, increasing propofol concentrations increases the inhibitory synaptic frequency response function \(\phi ^{i}_{ab}\) but the nonlinear gain functions \(S_{C,T}^{\prime }(V^{\ast e,i}_{b})\) decrease (cf. Fig. 10d). As observed in Fig. 11b, since no inhibitory synapses are involved in the ese loop, ψ e s e decreases with a decreasing nonlinear gain function. This reveals an important role for the ese loop in the modulation of α−activity which decreases by decreasing the gain function, in contrast to the upper resting state.
Within the eie, srs and e s r e loops, the nonlinear gains at the lower resting state decrease by an increase of the propofol concentration while the increase in the inhibitory charge transfer more than compensates this decrease essentially leading to an increase in ψ e i e , ψ s r s , and ψ e s r e . Consequently, in these loops the enhancement of inhibition plays a more substantial role than the gain function modulation.
3.3 Reduced model
The previous section considers both thalamic and cortical inhibition revealing their role in generating δ− and α−activity. Specifically, cortical inhibition is strong and balances cortical excitation. In order to study the role of the intra-thalamic inhibition for the generation of characteristic EEG pattern under anaesthesia sedation, we focus on the role of thalamic inhibition while neglecting inhibition within the cortex (although, of course, cortical inhibition is present in the brain) and refer to this model as reduced model in the following. This assumption simplifies the model under study, while the obtained results shall reveal that the reduced model is still adequate to reproduce observed changes in EEG rhythms within δ− and α−activity bands. The results show clearly that thalamic inhibition balances cortical excitation as cortical inhibition does. Figure 12 shows the power spectra of EEG over frontal and occipital regions in the baseline and in the anaesthetic conditions computed at the upper and lower resting states of the system. It turns out that the reduced model yields similar results compared to the full model (11), cf. Figs. 6 and 8.

The spectral power of EEG associated with the reduced thalamo-cortical model neglecting cortical inhibition in the baseline condition (p=1) and under sedation (p>1). The solid lines indicate the analytical solutions and the dashed lines show the numerical solutions of the model system. The panels show the spectrum computed at the upper and lower resting states in (a) and (b) reproducing the characteristics of experimental EEG spectra over the frontal and occipital regions, respectively. Parameters are taken from sets I, II in Table 1 while neglecting cortical inhibition
3.3.1 Frontal spectrum
Taking a closer look at the origin of the frontal EEG, Fig. 13a shows the spectral power of the system fluctuating about the upper resting state. It can be observed that under sedation (red line), the spectral power is well enhanced within the δ− and α−band in comparison to the baseline condition (blue line). Moreover, the δ−power peak emerges by increasing the concentration of propofol. Figure 13b and c give the relative contribution {w n } of the respective populations in the δ− and α−frequency bands, respectively. They show that the mean postsynaptic potentials (PSPs) \({V_{E}^{e}}\), \({V_{S}^{e}}\) and \({V_{S}^{i}}\) contribute most to the δ−activity while the excitatory PSPs involving in ese loop (i.e., \({V_{E}^{e}}\) and \({V_{S}^{e}}\)) generate the activity in α−band.

The spectral power and the partial contributions in the brain areas describing activity in the frontal electrodes. a Power spectrum in the baseline (p =1, blue) and sedation condition (p = 1.3, red). b Contributions w n (cf. Eq. (18)) in δ−band and c in the α−band. The system fluctuates about the upper resting state of the system. Parameters are α i = 200, β i = 20, and others are taken from set I in Table 1
To gain insight into the resting activity of the reduced model, we investigate the number of resting states of the system for possible combinations of different coupling connections. Figure 14 represents the parameter space where the system exhibits single and triple resting solutions, cf. Appendix C for the derivation of the resting state activity. It can be seen that the triple resting state occurs if the cortico-thalamic relay and relay-cortical connections are strong enough whereas the cortico-reticular and intra-thalamic connections are weak enough (cf. Fig. 14h, i and j). Moreover, cutting the connection between cortex and relay population leads to a single resting state independent of the other parameters.

Parameter space for different coupling connections. The shaded (unshaded) areas represent the parameter regions where the system exhibits triple (single) resting solutions. Parameters are taken from set I in Table 1
3.3.2 Occipital spectrum
To investigate the occipital EEG in some more detail, we consider different topological configurations to study δ− and α−activity. Figure 15 illustrates the power spectra and the relative contribution {w n }, generated by different anatomical circuit configurations computed at the lower resting state of the system in the baseline and anaesthetic conditions. At first, the plots reveal that the anaesthetic state always exhibits a diminished α−power compared to the baseline condition, when the thalamic inhibition is involved in the system.

Different topological configurations, their resulting spectral power and the contributions to power in the occipital electrodes. a Topology of the system. The solid and dashed arrows denote present and eliminated connections, respectively. b Spectral power in the baseline condition (p = 1, blue) and sedation condition (p = 1.3, red). c and d show contributions to power w n (cf. Eq. (18)) in the δ−band and α−band, respectively. The systems fluctuate about the lower resting state of the system. Parameters are taken from set II in Table 1
Figure 15a (I) shows a system with the simplest topology including a reciprocal projection between cortical pyramidal neurons (E) and thalamo-cortical relay neurons (S) associated with a time delay (ese circuit). This cortico-thalamic relay loop generates α−activity with a strong power spectral peak at about 10 Hz, as illustrated in Fig. 15b. The circuit also generates activity in the δ−band but no power spectral peak in this range. Figure 15c and d reveal that the spectral power is generated by the excitatory PSPs involving in this circuit, i.e., \({V_{E}^{e}}\) and \({V_{S}^{e}}\). Since no inhibitory synapses are involved here, the resulting spectrum is independent of the anaesthetic level.
Configurations (II) and (III) in Fig. 15a display the topology of the reduced model with the disrupted cortico-thalamic relay circuit. These configurations yield the loss of the prominent α−peak that indicate the important role of ese loop in the generation of α−activity. In configuration (II), we choose very small relay-cortical connection, because missing of this connection diminishes the power spectrum across the entire frequency bands (if K E S =0, then P E (ν)=0). In configuration (III), removing the cortico-thalamic relay connection retains the esre and srs circuits in the system that yield a strong oscillation at about 3 Hz with the clear corresponding spectral peak. In addition, Fig. 15c and d show that the PSPs \({V_{E}^{e}}\), \({V_{S}^{i}}\) and \({V_{R}^{e}}\) involved in these circuits generate the activity in δ−band.
Configuration (IV) demonstrates a system including the cortico-reticular, reticular-relay and relay-cortical connections (esre circuit). Similar to configuration (III), the resulting spectral power of this configuration exhibits a strong oscillation at about 3 Hz. Configurations (V) and (VI) illustrate the reduced model with a disrupted esre closed circuit. These configurations yield the loss of δ−peak but a strong α−power peak emerges, because the system comprises the ese circuit. This indicates the major contributions of esre and srs circuits to the generation of δ−activity and the ese circuit to generation of α−activity in the system.
To further reveal the role of esre and srs circuits in the generation of δ−activity and the ese circuit in the generation of α−activity, we investigate the effect of increasing connection strengths on the imaginary part (frequency) and real part (damping rate) of the characteristic root corresponding to the δ− and α−activity, respectively. Figure 16 illustrates that the frequency and the damping rate of δ−activity increase with increasing the cortico-reticular (K R E ), relay-cortical (K E S ) and intra-thalamic (K R S ,K S R ) connections indicating an increase of frequency and magnitude in the δ−band. In a similar manner, Fig. 17 shows that the frequency and the damping rate of α−activity increase with increasing the cortico-thalamic relay (K S E ) and relay-cortical (K E S ) connections which demonstrate the increasing of frequency and magnitude in the α−band. An additional study (not shown) of the remaining connections, i.e., relay-cortical (K E S ) connection in the generating of δ−activity and cortico-reticular (K R E ) and the intra-thalamic (K R S ,K S R ) connections in the generating of α−activity, reveals a decreasing effect on the generation of δ− and α−power, respectively. These results suggest the important role of esre and srs loops in the generation of δ−activity and ese loop in the generation of α−activity.

Modulation of the frequency and the corresponding damping rate of δ−activity by increase of coupling strengths in the esre and srs circuits. Parameters are taken from set II in Table 1

Modulation of the frequency and corresponding damping rate of the α−activity by increase of coupling strengths in the ese circuit. Parameters are taken from set II in Table 1
4 Discussion
4.1 Origin of spectral peaks
A large class of general anaesthetics such as propofol act on inhibitory GABAergic synaptic receptors and hence enhance the inhibition in the neural populations. Accordingly one might expect that the stronger inhibition caused by propofol would reduce spectral power. The opposite is in fact the case; stronger inhibition induces a power surge in certain frequency bands. Figures 7 and 9 explain this power surge as an approach of the system dynamics to dynamical oscillatory instabilities. In the control condition the external random fluctuations from other cortical and non-cortical areas are well damped except for the α−frequency band. The power surge of α−activity indicates that the system is lying in the vicinity of an oscillatory instability point. The nonlinear instabilities analysis in large-scale neural activity have been demonstrated in Breakspear et al. (2006) to explain the mechanisms underlying human generalized seizures. This explanation is in line with other studies investigating the origin of α−activity (Robinson et al. 2001a; Robinson et al. 2002; Drover et al. 2010; Hindriks and van Putten 2012; Hutt 2013). These models do not examine explicitly the effects of anaesthetic-induced prolongation of the IPSP. In our study, if the system is lying on the upper resting state, increasing the synaptic inhibition by increasing the anaesthetic concentration moves the system closer to the instability point, decreasing the damping of external fluctuations, and hence leading to an increase of power as observed in Fig. 6 for frontal electrodes. In contrast, if the system is lying on the lower branch increased inhibition moves the system away from the oscillatory instability point and hence a resultant decrease in power as observed in occipital electrodes, cf. Fig. 8.
To explain the origin of the power surge in the δ−frequency band, a slightly different mechanism comes into play since no spectral peak in the δ−range is present in the control condition. Hence increased synaptic inhibition may not only move the system towards an already existing instability point, but actually also generates this instability point for increased inhibition. Figures 7 and 9 explain the origin of the power surge in the δ−frequency band by the generation of an oscillatory instability at frequencies in the δ−band. A more detailed analysis (cf. Fig. 13) reveals that the δ−power surge is initiated at values p>1, i.e., does not occur in the control condition (p=1) but for increased anaesthetic concentration only. Hence, the spectral power peak in the δ−frequency band originates from the propofol-induced inhibition (Hashemi et al. 2014).
Our model results both extend and contrast previous findings. To the best of our knowledge, the model is one of the first taking into account the explicit anaesthetic action of propofol on thalamic GABAergic synaptic receptors, cf. Fig 3. The model suggests an explanation for the observed changes in the EEG rhythms in the δ− and the α−frequency bands and describes well the anteriorization of both the α− and δ−rhythms, i.e., the distinct dynamics in both frontal and occipital cortical regions.
It seems likely that the observed characteristic changes in EEG rhythms are not just simply signatures of cortical inhibition. Our results show that the changes observed experimentally in EEG during sedation can be reproduced with and without inhibitory cortical neurons, cf. Fig 12. The neglect of cortical inhibition put emphasis on the effect of propofol on the inhibitory synaptic receptors in the thalamic cells as well as reducing the dimensionality of the model. By reducing the dimensionality of the mathematical model we are able to utilize their analytical tractability to obtain some inequality conditions for the stability of the resting states and even to gain further insights into the mechanisms responsible for the spectral features (Hutt 2013). The application of analytical constraints on model parameters remains to be investigated in power spectral fitting methods, which it has the potential to improve previous work in fitting the model’s predictions to experimental data (Rowe et al. 2004; Robinson 2003; Robinson et al. 2004). This issue is beyond the scope of the present manuscript and it would be the aim of future work.
Moreover, it is important to note that such a reduced model does not rule out other proposed models elucidating the mechanisms underlying the effects of anaesthetic agents on brain dynamics. For instance, Hindriks and van Putten (2012) stressed the importance of cortical self-inhibition and supports our result that the increase of α−power over frontal region can be caused by increase in the gain functions of thalamocortical network. Our results reveal that it is possible to replicate several observed EEG phenomena by modeling anaesthetic action only in the thalamo-cortical loops. Several previous studies describing EEG spectra by a cortico-thalamic model consider both thalamic and cortical anaesthetic action (Ching et al. 2010; McCarthy et al. 2008; Hindriks and van Putten 2012; Vijayan et al. 2013) and point out the importance of cortical anaesthetic action. Even purely cortical models (Liley and Bojak 2005; Wilson et al. 2006) may explain several spectral features observed in EEG under anaesthesia. Most of these previous models are either rather complex with several tens of free parameters and hence many degrees of freedom permitting to fit the experimental data for a small subset of parameters (Liley and Bojak 2005; Wilson et al. 2006) or they identify specific single neuron mechanisms to be responsible for a certain anaesthetic effect in macroscopic EEG, e.g., as in Ching et al. (2010), Vijayan et al. (2013), and McCarthy et al. (2008). In contrast, our model is comparatively simple while proposing a major underlying mechanism: the activation of pre-defined oscillation modes whose amplitude and frequency depends on the level of synaptic inhibition.
The present work considers only light sedation, i.e., low propofol concentrations, far from those required for full surgical anaesthesia. Some previous studies have investigated in detail the EEG power spectra at higher concentrations of propofol where additional characteristic features emerge, such as burst suppression (Ching et al. 2012), broad spatial EEG coherence (Cimenser et al. 2011; Murphy et al. 2011; Gugino et al. 2001; Purdon et al. 2013), strong spectral power in the β−band (McCarthy et al. 2008; Purdon et al. 2013) and functional fragmentation accompanied by a drop of population firing rate and increased spectral power at frequencies <1 Hz (Lewis et al. 2012). For instance, Fig. 5c shows emerging β−activity well after light sedation that we consider in the present work. Our model does not explain these latter features, since they emerge for higher propofol concentrations and may result from fundamental changes in the interaction between neural structures, cf. Ching et al. (2010) explaining the β−power surge by the interaction of two thalamo-cortical modules. To describe the features seen at high anaesthetic concentration, it is necessary to incorporate more features of anaesthetic actions on neural population than the effects considered in this study. Some of our preliminary simulations (not shown) demonstrate that, by assuming that anaesthesia delays axonal transmission, the model used in this study is able to reproduce the β−power surge. However, this effect on the axonal transmission delay needs more experimental indication. Furthermore, recent studies have linked the low-frequency phase modulation of the alpha oscillation amplitudes to different states of unconsciousness (Purdon et al. 2013; Mukamel et al. 2014). They have reported that two distinct patterns can be observed during propofol anaesthesia. First, the trough-max pattern which appears at the transition into and out of unconsciousness. Second, the peak-max pattern which appears at profound unconsciousness. While these patterns can be used to track the states of unconsciousness, however, the mechanisms underlying these patterns remain to be understood. Such phase-amplitude modulation patterns are not visible in power spectral analysis and we postpone their discussion to later work since the corresponding discussion would exceed the major aim of the present work.
Our model describes very well the action of propofol on neural populations in the cortex and thalamus while the fundamental interactions of cortex, thalamus and other sub-cortical structures such as the brainstem and hippocampus are retained. In contrast, larger anaesthetic concentrations may affect not only the activity in single populations but also the interaction between a larger set of neural structures. This effect on the inter-area interactions has been been quantified in several previous experimental studies and has been termed fragmentation or connectivity break-down (Lewis et al. 2012; Boly et al. 2012; Massimini et al. 2005).
4.2 Multiple resting states
Experimental EEG under propofol anaesthesia shows an anteriorization in the δ− and α−frequency bands, cf. Fig. 5 and Purdon et al. (2013), Cimenser et al. (2011), and Lewis et al. (2012). The recent study of Vijayan et al. (2013) is one of the first to explain the distinct evolution of spectral power. They suggest two distinct models for the anteriorization during propofol anaesthesia: one for the frontal and one for the occipital EEG α−activity; where the occipital cortico-thalamic connection exhibits an additional hyperpolarizing mechanism lowering the resting state population firing rate. Their model is based on simulations of small numbers of neurons, each with multiple specialised ion channels. The anteriorization is largely driven by a specialised subset of thalamic relay neurons that project only to the occipital cortex. The oscillatory activity of these neurons secondarily induce high- and low-activity resting states. In our study, different systems with distinct parameter sets explain the spectral power of EEG associated with frontal and occipital head regions. In addition, our model extends the explanation of Vijayan et al. (2013) by providing a more generic dynamical systems explanation for both the δ− and the α−frequency bands, and emphasizes the importance of nonlinear gain functions in the corresponding resting states, rather than molecular scale ion channel activity. Our mathematical analysis of the power spectrum (15) reveals that these gain functions are dependent on the resting potential values. Increasing nonlinear cortical gain functions induce a power surge in the α−range, whereas decreasing nonlinear cortical gain functions diminish the α−power. This is in line with the cortical activation hypothesis, which states that an increase or decrease in the firing rates of cortical pyramidal neurons leads to an increase or decrease in α−activity, respectively (Hashemi et al. 2014; Hindriks and van Putten 2013; Rowe et al. 2004). Since increases in the nonlinear gains occur at high activity-resting states only, while decreasing nonlinear gains are present in low activity-resting states (Fig. 10), our findings suggest the important role of multiple resting states at high and low population firing rates and affirms previous EEG modeling studies (Steyn-Ross et al. 2001b; Victor et al. 2011; Drover et al. 2010) pointing out the importance of multiple resting states to explain unstimulated EEG. This is affirmed by Fig. 14 illustrating that cutting the connections between cortex and relay population leads the loss of multiple states and consequently the destruction of realistic frontal population activity. In contrast, the evolution of the δ−power is rather independent of the resting state, and increases for increasing propofol concentrations for both increasing and decreasing nonlinear gains. This strongly suggests that the α−rhythm is fundamentally different from the δ−rhythm.
4.3 Effective sub-circuits in the cortico-thalamic model
As argued before, the origin of the δ−rhythm appears to be fundamentally different to the origin of α−rhythm. A detailed analysis of the spectral contributions of the populations to δ− and α−power, cf. Figs. 13 and 15, reveals that strong δ−power occurs primarily when there is a strong connection between thalamic areas (srs loop) and from cortex to the reticular nucleus, reticular to relay nucleus and relay to cortex (esre loop), while strong α−power is generated primarily by the cortico-thalamic relay circuit (ese loop). These results are based on an investigation of small deviations from the resting state in light sedation and are not directly related to nonlinear signal features observed in deeper anaesthesia states, such as spindles.
Having identified these sub-circuits, their subsequent detailed analysis reveals that the frontal enhancement and occipital attenuation of α−activity originate primarily from a (frontal) increase and (occipital) decrease in the nonlinear gain functions within the thalamo-cortical ese loop. In turn, this result supports the presence of multiple resting states derived in the earlier part of our analysis. Hence, we suggest that the phenomenon of anteriorization of α−power results from fronto-occipital differences in the nonlinear gain functions in the cortico-thalamic relay circuit. The previous study of Vijayan et al. (2013) on the anteriorization of α−activity supports a similar idea. These results are also in agreement with the recent findings of Hindriks and van Putten (2013). They show that changes of transfer function properties and synaptic parameters, directly or indirectly, lead to changes in the gain functions by altering the values of the resting states of the system that cause the α−response modulations. Their results affirm our hypothesis that the increase or decrease of α−power spectrum depends profoundly on the the resting state values of the system and the corresponding nonlinear gain functions. This finding is well in line with previous modeling studies on the origin of occipital α−activity in the absence of anaesthesia explaining the amplitude modification by modified nonlinear gain changes.
The changes in the δ−frequency band are explained by increases in the charge transfer within the inhibitory synapses what results to an increase of the frequency response function within the loops involving inhibitory neurons (such as srs and esre loops), which essentially enhances the δ−power. This hypothesis on the interplay of nonlinear gain and synaptic inhibition has been briefly mentioned in a previous work (Hutt 2013).
Our analysis aims to extract general interaction mechanism for the generation of spectral EEG characteristics. To this end, we employ a reductionist approach: starting from a complex model and reducing it step by step to find a neurophysiologically reasonable minimal model that explains as much experimental features as possible. The approach is motivated by the insight that general anaesthesia is a macroscopic global and fundamental effect that occurs in all species of animals, even though they exhibit different detailed neural structures. Consequently, the underlying mechanism should be rather unspecific in biological detail but rather general in concept. We have identified elements of principal underlying mechanisms to explain δ− and α−power observed during propofol anaesthesia sedation in adult humans, namely the nonlinear population gain, the level of synaptic inhibition and the interaction of neural sub-circuits (or microcircuits). It appears that one of the most important properties of the sub-circuits is their ability to generate an oscillation. Since these elements promise to reflect underlying principles, they are rather independent of the choice of the anaesthetic agent and may generalize to the larger class of anaesthetic GABAergic (Garcia et al. 2010) or volatile drugs (Grasshoff et al. 2006).
Several previous modeling studies have explained the α−rhythm by the delayed thalamo-cortical feedback (Robinson et al. 2001a; Robinson et al. 2002; Rennie et al. 2002; Chiang et al. 2008; Hindriks and van Putten 2013) or by a cortical model, cf. e.g., Spiegler et al. (2010), Spiegler et al. (2011), and Hutt (2013). At first glance these two explanations appear exclusive. However the underlying principle put forward in the present work is not contradictory to cortical and thalamo-cortical circuits since both represent oscillating sub-circuits. Previous cortical models have included feedback loops of excitatory and inhibitory populations and most thalamo-cortical feedback models oscillate due to delay, which is known to facilitate oscillations (Robinson et al. 2001b; Rowe et al. 2004). Consequently, the α−rhythm may originates from a general class of oscillating circuits. This interpretation is supported by the experimental findings that the brain is highly adaptive and brain areas may take over tasks of other areas as seen under general anaesthesia (Lee et al. 2010), in sleep (Siegel 2009) or after brain injury (Wieloch and Nikolich 2006).
Similarly, various sources and mechanisms have been proposed for EEG δ−activity. Previous experimental studies suggest a cortical origin of δ−waves under anaesthesia (Murphy et al. 2011) and point out the importance of the cortico-thalamic feedback in the generation of δ−waves in anaesthesia (Boly et al. 2012) and sleep (Steriade et al. 1993; Dang-Vu et al. 2008), while δ−activity during sleep may also results from a connected network of thalamus, brainstem and different cortical areas (Maquet et al. 1997). Our model results point out the importance of the intra-thalamic feedback of reticular and relay populations as part of the thalamo-cortical feedback loop, which is in full line with the previous literature.
4.4 Model limitations
Our model is concentrates on the effects of low concentrations of propofol. Additional spectral EEG features, such as an increase in β−power just before loss of consciousness (Purdon et al. 2013), or burst suppression patterns (Ching et al. 2012; Liley and Walsh 2013) which emerge at high levels of anaesthetic concentrations, remain to be investigated in the future work. To explain these features, a future model will incorporate additional thalamo-cortical modules and nonlinear dynamics in thalamic populations. In addition, the model fitting with experimental data will reveal the optimal topology model that is able to reproduce the observed specific features in experimental data (Friston et al. 2003).
Moreover, the model neglects additional, possibly important, anaesthetic effects such as the tonic inhibition induced at extra-synaptic GABAergic receptors (Farrant and Nusser 2005; Hutt 2012) which are very sensitive to anaesthetics (Orser 2006) and are supposed to determine the global level of inhibition (Semyanov et al. 2004; Belelli et al. 2009). A recent work on the effect of extra-synaptic anaesthetic action on neural populations (Hashemi et al. 2014; Hutt and Buhry 2014) has elucidated how to incorporate this effect in the model presented in this work. In addition, extra-synaptic receptors are found in several sub-cortical areas (Kullmann et al. 2005; Farrant and Nusser 2005) indicating a strong effect on the reticular activating system (RAS) (Vanini and Baghdoyan 2013). Hence, to understand better the inhibitory homeostatic mechanisms in anaesthesia, it will be necessary to include the RAS and its involved sub-cortical areas.
5 Conclusion
We would conclude that - using a model of cortico-thalamic loops - it is possible to replicate the different frontal and occipital changes in the α− and δ−rhythms in the EEG, caused by modest concentrations of propofol. The α−rhythm is primarily dependent on the cortico-thalamic relay interactions; dependent on the mean potential values of the resting states of the system, an increase or decrease in the gain functions within the thalamo-cortical circuits results in an increase or decrease in the spectral power in the α−band, whereas the increased propofol inhibition acting via the thalamo-cortical loops is necessary for the increase in δ−waves. This model also offers a plausible explanation of the eyes-closed occipital alpha rhythm.
References
Alkire, M.T., Haier, R.J., & Fallon, J.H. (2000). Toward a unified theory of narcosis: brain imaging evidence for a thalamocortical switch as the neurophysiologic basis of anesthetic-induced unconsciousness. Consciousness and Cognition, 9, 370–386.
Alkire, M.T., Hudetz, A.G., & Tononi, G. (2008). Consciousness and anesthesia. Science, 322, 876–880. doi:10.1126/science.1149213.
Amari, S. (1977). Dynamics of pattern formation in lateral-inhibition type neural fields. Biological Cybernetics, 27, 77–87.
Antkowiak, B. (2002). In vitro networks: cortical mechanisms of anaesthetic action. British Journal of Anaesthesia, 89(1), 102–111.
Belelli, D., Harrison, N.L., Maguire, J., Macdonald, R.L., Walker, M.C., & Cope, D.W. (2009). Extra-synaptic GABA A receptors: form, pharmacology, and function. The Journal of Neuroscience, 29(41), 12757–12763.
Bojak, I., & Liley, D.T.J. (2005). Modeling the effects of anesthesia on the electroencephalogram. Physical Review E, 71, 041902.
Boly, M., Moran, R., Murphy, M., Boveroux, P., Bruno, M.A., Noirhomme, Q., Ledoux, D., Bonhomme, V., Brichant, J.F., Tononi, G., Laureys, S., & Friston, K.I. (2012). Connectivity changes underlying spectral EEG changes during propofol-induced loss of consciousness. The Journal of Neuroscience, 32(20), 7082–7090.
Breakspear, M., Roberts, J., Terry, J.R., Rodrigues, S., Mahant, N., & Robinson, P.A. (2006). A unifying explanation of primary generalized seizures through nonlinear brain modeling and bifurcation analysis. Cerebral Cortex, 16, 1296–1313.
Bressloff, P. (2012). Spatiotemporal dynamics of continuum neural fields. Journal of Physics A, 45, 033001.
Byoung-Kyong, M. (2010). A thalamic reticular networking model of consciousness. Theoretical Biology and Medical Modelling, 7.
Chau, P-L. (2010). New insights into the molecular mechanisms of general anaesthetics. British Journal of Pharmacology, 161, 288–307.
Chiang, A.K., Rennie, C.J., Robinson, P.A., Roberts, J.A., Rigozzi, M.K., Whitehouse, R.W., Hamilton, R.J., & Gordon, E. (2008). Automated characterization of multiple alpha peaks in multi-site electroencephalograms. Journal of Neuroscience Methods, 168, 396–411.
Ching, S., Cimenser, A., Purdon, P.L., Brown, E.N., & Kopell, N.J. (2010). Thalamocortical model for a propofol-induced-rhythm associated with loss of consciousness. Proceedings of the National Academy of Sciences of the United States of America, 107(52), 22665–22670.
Ching, S., Purdon, P.L., Vijayand, S., Kopell, N.J., & Brown, E.N. (2012). A neurophysiological metabolic model for burst suppression. Proceedings of the National Academy of Sciences of the United States of America, 109 (8), 3095–3100.
Cimenser, A., Purdon, P.L., Pierce, E.T., Walsh, J.L., Salazar-Gomez, A.F., Harrell, P.G., Tavares-Stoeckel, C., Habeeb, K., & Brown, E.N. (2011). Tracking brain states under general anesthesia by using global coherence analysis. Proceedings of the National Academy of Sciences of the United States of America, 108(21), 8832–8837.
Dang-Vu, T.T., Schabus, M., Desseilles, M., Albouy, G., Boly, M., Darsaud, A., Gais, S., Rauchs, G., Sterpenich, V., Vandewalle, G., Carrier, J., Moonen, G., Balteau, E., Degueldre, C., Luxen, A., Phillips, C., & Maquet, P. (2008). Spontaneous neural activity during human slow wave sleep. Proceedings of the National Academy of Sciences of the United States of America, 105(39), 15160–15165.
David, O., & Friston, K.J. (2003). A neural mass model for meg/eeg: coupling and neuronal dynamics. NeuroImage, 20, 1743– 1755.
David, O., Kiebel, S.J., Harrison, L.M., Mattout, J., Kilner, J.M., & Friston, K.J. (2006). Dynamic causal modeling of evoked responses in eeg and meg. NeuroImage, 30, 1255–1272.
Deco, G., Jirsa, V.K., Robinson, P.A., Breakspear, M., & Friston, K. (2008). The dynamic brain: from spiking neurons to neural masses and cortical fields. PLoS Computational Biology, 4(8).
Drover, J.D., Schiff, N.D., & Victor, J.D. (2010). Dynamics of coupled thalamocortical modules. Journal of Computational Neuroscience, 28, 605–616.
Farrant, M., & Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA A receptors. Nature Reviews Neuroscience, 6, 215–229.
Feshchenko, V.A., Veselis, R.A., & Reinsel, R.A. (2004). Propofol-induced alpha rhythm. Neuropsychobiology, 50(3), 257–266.
Foster, B.L., Bojak, I., & Liley, D.T.J. (2008). Population based models of cortical drug response: insights from anaesthesia. Cognitive Neurodynamics, 2, 283–296.
Franks, N.P. (2008). General anesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nature Reviews Neuroscience, 9, 370–386. doi:10.1038/nrn2372.
Franks, N.P., & Lieb, W.R. (1994). Molecular and cellular mechanisms of general anesthesia. Nature, 367, 607–614.
Freeman, W.J. (1979). Nonlinear gain mediating cortical stimulus-response relations. Biological Cybernetics, 33, 237–247.
Freestone, D.R., Aram, P., Dewar, M., Scerri, K., Grayden, D.B., & Kadirkamanathan, V. (2011). A data-driven framework for neural field modeling. NeuroImage, 56(3), 1043–1058.
Friston, K.J., Harrison, L., & Penny, W. (2003). Dynamic causal modelling. NeuroImage, 19, 273–1302.
Garcia, P.S., Kolesky, S.E., & Jenkins, A. (2010). General anesthetic actions on GABA A receptors. Current Neuropharmacology, 8(1), 2–9.
Grasshoff, C., Drexler, B., Rudolph, U., & Antkowiak, B. (2006). Anaesthetic drugs: linking molecular actions to clinical effects. Current Pharmaceutical Design, 12(28), 3665–3679.
Gugino, L.D., Chabot, R.J., Prichep, L.S., John, E.R., Formanek, V., & Aglio, L.S. (2001). Quantitative EEG changes associated with loss and return of conscious- ness in healthy adult volunteers anaesthetized with propofol or sevoflurane. British Journal of Anaesthesia, 87, 421–428.
Hashemi, M., Hutt, A., & Sleigh, J. (2014). Anesthetic action on extra-synaptic receptors: effects in neural population models of EEG activity. Journals of Frontiers in Systems Neuroscience, 8(232).
Hazeaux, C., Tisserant, D., Vespignani, H., Hummer-Sigiel, L., Kwan-Ning, V., & Laxenaire, M.C. (1987). Electroencephalographic changes produced by propofol. Annales Françaises d’Anesthèsie et de Rèanimation, 6, 261–266.
Hindriks, R., & van Putten, M.J.A.M. (2012). Meanfield modeling of propofol-induced changes in spontaneous EEG rhythms. Neuroimage, 60, 2323–2344.
Hindriks, R., & van Putten, M.J.A.M. (2013). Thalamo-cortical mechanisms underlying changes in amplitude and frequency of human alpha oscillations. Neuroimage, 70, 150–163.
Hutt, A. (2012). The population firing rate in the presence of GABAergic tonic inhibition in single neurons and application to general anaesthesia. Cognitive Neurodynamics, 6, 227–237.
Hutt, A. (2013). The anaesthetic propofol shifts the frequency of maximum spectral power in EEG during general anaesthesia: analytical insights from a linear model. Frontiers in Computational Neuroscience, 7, 2.
Hutt, A., & Buhry, L. (2014). Study of GABAergic extra-synaptic tonic inhibition in single neurons and neural populations by traversing neural scales: application to propofol-induced anaesthesia. Journal of Computational Neuroscience. in press.
Hutt, A., & Longtin, A. (2009). Effects of the anesthetic agent propofol on neural populations. Cognitive Neurodynamics, 4(1), 37–59.
Hutt, A., Bestehorn, M., & Wennekers, T. (2003). Pattern formation in intracortical neuronal fields. Network: Computation in Neural Systems, 14, 351–368.
Jirsa, V.K., & Haken, H. (1996). Field theory of electromagnetic brain activity. Physical Review Letters, 77 (5), 960–963.
Johnson, B.W., Sleigh, J.W., Kirk, I.J., & Williams, M.L. (2003). High-density EEG mapping during general anaesthesia with xenon and propofol: a pilot study. Anaesthesia and Intensive Care, 31(2), 155–163.
Kitamura, A., Marszalec, W., Yeh, J.Z., & Narahashi, T. (2002). Effects of halothane and propofol on excitatory and inhibitory synaptic transmission in rat cortical neurons. Journal de Pharmacologie, 304(1), 162–171.
Kullmann, D.M., Ruiz, A., Rusakov, D.M., Scott, R., Semyanov, A., & Walker, M.C. (2005). Presynaptic, extrasynaptic and axonal GABA A receptors in the cns: where and why?. Progress in Biophysics and Molecular Biology, 87, 33–46.
Lee, U., Oh, G., Kim, S., Noh, G., Choi, B., & Mashour, G.A. (2010). Brain networks maintain a scale-free organization across consciousness, anesthesia, and recovery: Evidence for adaptive reconfiguration. Anesthesiology, 113(5), 1081–1091.
Lewis, L.D., Weiner, V.S., Mukamel, E.A., Donoghue, J.A., Eskandar, E.N., Madsen, J.R., Anderson, W.S., Hochberg, L.R., Cash, S.S., Brown, E.N., & Purdon, P.L. (2012). Rapid fragmentation of neuronal networks at the onset of propofol-induced unconsciousness. Proceedings of the National Academy of Sciences of the United States of America, 109(21), 3377–3386.
Liley, D.T.J., & Bojak, I. (2005). Understanding the transition to seizure by modeling the epileptiform activity of general anaesthetic agents. Journal of Clinical Neurophysiology, 22, 300–313.
Liley, D.T.J., & Walsh, M. (2013). The mesoscopic modeling of burst suppression during anesthesia. Frontiers in Computational Neuroscience, 7, 46.
Liley, D.T.J., & Wright, J.J. (1994). Intracortical connectivity of pyramidal and stellate cells: estimates of synaptic densities and coupling symmetry. Network: Computation in Neural Systems, 5, 175–189.
Liley, D.T.J., Cadusch, P.J., & Dafilis, M.P. (2002). A spatially continuous mean field theory of electrocortical activity. Network: Computation in Neural Systems, 13, 67–113.
Maquet, P., Degueldre, C., Delfiore, G., Aerts, J., Peters, J., Luxen, A., & Franck, G. (1997). Functional neuroanatomy of human slow wave sleep. The Journal of Neuroscience, 17(8), 2807–2812.
Massimini, M., Ferrarelli, F., Huber, R., Esser, S.K., Singh, H., & Tononi, G. (2005). Breakdown of cortical effective connectivity during sleep. Science, 309, 2228–2232.
McCarthy, M.M., Brown, E.N., & Kopell, N. (2008). Potential network mechanisms mediating electroencephalographic beta rhythm changes during propofol-induced paradoxical excitation. The Journal of Neuroscience, 28(50), 13488–13504.
Molaee-Ardekani, B., Senhadji, L., Shamsollahi, M.B., Vosoughi-Vahdat, B., & Wodey, E. (2007). Brain activity modeling in general anesthesia: Enhancing local mean-field models using a slow adaptive firing rate. Physical Review E, 76, 041911.
Mukamel, E.A., Pirondini, E., Babadi, B., Wong, K.F.k., Pierce, E.T., Harrell, P.G., Walsh, J.L., Salazar-Gomez, A.F., Cash, S.S., Eskandar, E.N., Weiner, V.S., Brown, E.N., & Purdon, P.L. (2014). A transition in brain state during propofol-induced unconsciousness. Journal of Neuroscience, 34(3), 839–845.
Murphy, M., Bruno, M.A., Riedner, B.A., Boveroux, P., Noirhomme, Q., Landsness, E.C., Brichant, J.F., Phillips, C., Massimini, M., Laureys, S., Tononi, G., & Boly, M. (2011). Propofol anesthesia and sleep: A high-density EEG study. Sleep, 34(3), 283–291.
Nunez, P.L. (1974). The brain wave equation: A model for the EEG. Mathematical Biosciences, 21, 279–291.
Nunez, P.L. (1981). Electrical fields of the brain. Oxford: Oxford University Press.
Nunez, P.L., & Srinivasan, R. (2006). Electric fields of the brain: The neurophysics of EEG. New York - Oxford: Oxford University Press.
Orser, B.A. (2006). Extrasynaptic GABA A receptors are critical targets for sedative-hypnotic drugs. Journal of Clinical Sleep Medicine, 2, 12–8.
Pinotsis, D.A., Moran, R.J., & Friston, K.J. (2012). Dynamic causal modeling with neural fields. NeuroImage, 59(2), 1261–1274.
Purdon, P.L., Pierce, E.T., Mukamel, E.A., Prerau, M.J., Walsh, J.L., Wong, K.F., Salazar-Gomez, A.F., Harrell, P.G., Sampson, A.L., Cimenser, A., Ching, S., Kopell, N.J., Tavares-Stoeckel, C., Habeeb, K., Merhar, R., & Brown, E.N. (2013). Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proceedings of the National Academy of Sciences of the United States of America, 110 (12), 1142–1151.
Reed, M.D., Yamashita, T.S., Marx, C.M., Myers, C.M., & Blumer, J.L. (1996). A pharmacokinetically based propofol dosing strategy for sedation of the critically ill, mechanically ventilated pediatric patient. Critical Care Medicine, 24(9), 1473–1481.
Rennie, C.J., Robinson, P.A., & Wright, J.J. (2002). Unified neurophysical model of EEG spectra and evoked potentials. Biological Cybernetics, 86, 457–471.
Robinson, P.A. (2003). Neurophysical theory of coherence and correlations of electroencephalographic signals. Journal of Theoretical Biology, 222, 163–175.
Robinson, P.A., Loxley, P.N., O’Connor, S.C., & Rennie, C.J. (2001b). Modal analysis of corticothalamic dynamics, electroencephalographic spectra, and evoked potentials. Physical Review E, 63, 041909.
Robinson, P.A., Rennie, C.J., & Rowe, D.L. (2002). Dynamics of large-scale brain activity in normal arousal states and eplieptic seizures. Physical Review E, 65(4), 041924.
Robinson, P.A., Rennie, C.J., & Wright, J.J. (1997). Propagation and stability of waves of electrical activity in the cerebral cortex. Physical Review E, 56, 826–840.
Robinson, P.A., Wright, J.J., & Rennie, C.J. (1998). Synchronous oscillations in the cerebral cortex. Physical Review E, 57, 4578–4588.
Robinson, P.A., Rennie, C.J., Wright, J.J., Bahramali, H., Gordon, E., & Rowe, D. (2001a). Prediction of electroencephalographic spectra from neurophysiology. Physical Review E, 63, 201903.
Robinson, P.A., Rennie, C.J., Rowe, D.L., & O’Connor, S.C. (2004). Estimation of multiscale neurophysiologic parameters by electroencephalographic means. Human Brain Mapping, 23, 53–72.
Rowe, D.L, Robinson, P.A., & Rennie, C.J. (2004). Estimation of neurophysiological parameters from the waking EEG using a biophysical model of brain dynamics. Journal of Theoretical Biology, 231(3), 413–433.
Rudolph, U., & Antkowiak, B. (2004). Molecular and neuronal substrates for general anaesthetics. Nature Reviews Neuroscience, 5, 709–720.
San-Juan, D., Chiappa, K.H., & Cole, A.J. (2010). Propofol and the electroencephalogram. Clinical Neurophysiology, 121(7), 998–1006.
Sellers, K.K., Bennett, D.V., Hutt, A., & Frohlich, F. (2013). Anesthesia differentially modulates spontaneous network dynamics by cortical area and layer. Journal of Neurophysiology. in press.
Semyanov, A., Walker, M.C., Kullmann, D.M., & Silver, R.A. (2004). Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends in Neurosciences, 27(5), 262–269.
Siegel, J.M. (2009). Sleep viewed as a state of adaptive inactivity. Nature. Reviews in the Neurosciences, 10, 747–753.
Spiegler, A., Kiebel, S.J., Atay, F.M., & Knosche, T.R. (2010). Bifurcation analysis of neural mass models: Impact of extrinsic inputs and dendritic time constants. NeuroImage, 52(3), 1041–1058.
Spiegler, A., Knosche, T.R., Schwab, K., Haueisen, J., & Atay, F.M. (2011). Modeling brain resonance phenomena using a neural mass model. PLoS Computational Biology, 7(12), 1002298.
Steriade, M., Contreras, D., Curro Dossi, R., & Nunez, A. (1993). The slow (< 1 hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. The Journal of Neuroscience, 13(8), 3284–3299.
Steyn-Ross, M.L., Steyn-Ross, D.A., & Sleigh, J.W. (2004). Modelling general anaesthesia as a first-order phase transition in the cortex. Progress in Biophysics and Molecular Biology, 85(2-3), 369– 385.
Steyn-Ross, M.L., Steyn-Ross, D.A., Sleigh, J.W., & Liley, D.T.J. (1999). Theoretical electroencephalogram stationary spectrum for a white-noise-driven cortex: Evidence for a general anesthetic-induced phase transition. Physical Review E, 60(6), 7299–7311.
Steyn-Ross, M.L., Steyn-Ross, D.A., Sleigh, J.W., & Wilcocks, L.C. (2001a). Toward a theory of the general-anesthetic-induced phase transition of the cerebral cortex: I. a thermodynamic analogy. Physical Review E, 64, 011917.
Steyn-Ross, M.L., Steyn-Ross, D.A., Sleigh, J.W., & Wilcocks, L.C. (2001b). Toward a theory of the general-anesthetic-induced phase transition of the cerebral cortex: Ii. numerical simulations, spectra entropy, and correlation times. Physical Review E, 64, 011918.
Vanini, G., & Baghdoyan, H.A. (2013). Extrasynaptic GABA A receptors in rat pontine reticular formation increase wakefulness. Sleep, 36(3), 337–343.
Victor, J.D., Drover, J.D., Conte, M.M., & Schiff, N.D. (2011). Mean-field modeling of thalamocortical dynamics and a model-driven approach to EEG analysis. Proceedings of the National Academy of Sciences of the United States of America, 108, 15631– 15638.
Vijayan, S., Ching, S., Cimenser, A., Purdon, P.L., Brown, E.N., & Kopell, N.J. (2013). Thalamocortical mechanisms for the anteriorization of alpha rhythms during propofol-induced unconsciousness. The Journal of Neuroscience, 33(27), 11070–11075.
Wieloch, T., & Nikolich, K. (2006). Mechanisms of neural plasticity following brain injury. Current Opinion in Neurobiology, 16(3), 258–264.
Wilson, H.R., & Cowan, J.D. (1972). Excitatory and inhibitory interactions in localized populations of model neurons. Biophysical Journal, 12, 1–24.
Wilson, M.T., Sleigh, J.W., Steyn-Ross, A., & Steyn-Ross, M.L. (2006). General anesthetic-induced seizures can be explained by a mean-field model of cortical dynamics. Anesthesiology, 104(3), 588–593.
Wright, J.J., & Liley, D.T.J. (1996). Dynamics of the brain at global and microscopic scales: Neural networks and the EEG. Behavioral and Brain Sciences, 19, 285–320.
Ying, S., & Goldstein, P. (2001). Propofol effects on the thalamus: Modulation of GABAergic synaptic inhibition and suppression of neuronal excitability. Abstract Viewer/Itinerary Planner Washington. DC: Society for Neuroscience, 89(411).
Ying, S.W., & Goldstein, P.A. (2005a). Propofol-block of SK channels in reticular thalamic neurons enhances GABAergic inhibition in relay neurons. Journal of Neurophysiology, 93, 1935–1948.
Ying, S.W., & Goldstein, P.A. (2005b). Propofol suppresses synaptic responsiveness of somatosensory relay neurons to excitatory input by potentiating GABA A receptor chloride channels. Molecular Pain, 1, 2.
Zhang, M., Wei, G.W., & Wei, C.H. (2002). Transition from intermittency to periodicity in lag synchronization in coupled roessler oscillators. Physical Review E, 65, 036202.
Zhou, C., Liu, J., & Chen, X.D. (2012). General anesthesia mediated by effects on ion channels. World Journal of Critical Care Medicine, 1, 80–93.
Acknowledgments
The authors acknowledge funding from the European Research Council for support under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 257253.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interests
The authors declare that they have no conflict of interest.
Appendices
Appendix A: Stationary states
Under the assumption of the spatial homogeneity, the mean excitatory and inhibitory postsynaptic potentials in the cortical pyramidal neurons (E), cortical inhibitory neurons (I), thalamo-cortical relay neurons (S) and thalamic reticular neurons (R) shown in Fig. 1 obey
In this model, the long-range propagation of the signals has been considered by the connection between cortex and thalamus associated with a constant time delay and the model follows closely Victor et al. (2011) and Drover et al. (2010) showing that long connections between cortico-cortical populations through white matter are not necessary to describe experimental EEG dynamics. In the case of constant external input I(t)=I 0, the spatially-homogeneous resting states of Eqs. (19) can be obtained by \(dV^{e,i}_{a}/dt=0\), for a∈{E,I,S,R} leading to
The dynamic of the reduced model are described by
where the resting states of the system obey
By inserting these equations into each other
where \(H[V^{*e}_{E}] \equiv {K_{RE} S_{C}[V^{*e}_{E}]+\frac {K_{RS}}{K_{ES}} V^{*e}_{E}} \). Since all the resting states \(V^{*e,i}_{a}\) for a∈{E,R,S} can be written as an implicit function of \(V^{*e}_{E}\), the number of solutions of \(V^{*e}_{E}\), i.e., the number of roots of Eq. (23), is identical to the number of resting states (Robinson et al. 1997; Robinson et al. 1998; Robinson et al. 2004).
Appendix B: Theoretical power spectrum
The solution of Eq. (14) for t→∞ is
with the matrix Greens function \(\boldsymbol {G}\in {\mathbb R}^{N\times N}\), that has dimension N = 7. Substituting Eq. (24) into Eq. (14) leads to
with the unitary matrix \(\boldsymbol {1} \in \mathbb {R}^{N \times N}\). Then the Fourier transform of the matrix Greens function
and the Wiener-Khinchine theorem defines the power spectral density matrix to
where the high index ⊤ denotes the transposed vector or matrix. Essentially we assume that the EEG is generated by the activity of pyramidal cortical cells. By virtue of the specific choice of external input to relay neurons, the power spectrum of the EEG just depends on one matrix component of the Greens function by
In detail, we find
and the diagonal matrix \(\hat {\boldsymbol {L}} (\partial /\partial t,p)\) with the entries \(\hat {L}_{1,1} = \hat {L}_{3,3} = \hat {L}_{5,5} = \hat {L}_{7,7}=\hat {L}_{e}(\nu )\), \(\hat {L}_{2,2} =\hat {L}_{4,4}=\hat {L}_{6,6} = \hat {L}_{i}(\nu ,p)\) and
with
The constants K i , i=1,…,9 depend on the anaesthetic factor p since they are evaluated at the resting state depending on p itself. Hence
and finally
Appendix C: Contribution to power spectrum
Here we consider the reduced model and parametrize the contribution of PSPs to the power spectrum in the δ− and α− frequency bands. Substituting the ansatz Y(t)=e λt u with eigenfunction \(\mathbf {u}=[ {u^{e}_{E}}, {u^{e}_{S}}, {u^{i}_{S}}, {u^{e}_{R}}]^{\top } \) into Eq. (14) yields
Now we can write all the elements of eigenfunction u in terms of the first element \({u^{e}_{E}}\) as follows
Then the associated normalized eigenfunction with known eigenvalue λ is \(\hat {\mathbf {u}} = C\left (1,u_{2}(\lambda ),u_{3}(\lambda ),u_{4}(\lambda )\right )^{\top }\), where \(C=\frac {1}{\sqrt {1+{u^{2}_{2}}+{u^{2}_{3}}+{u^{2}_{4}}}}\). All elements of vector Y(t)=[y 1(t),y 2(t),y 3(t),y 4(t)]⊤ can be written as \(y_{n}(t)=\hat u_{n} e^{\lambda t}+ \hat {u}_{n}^{\ast } e^{\lambda ^{\ast } t}\) for n=1,...,4. Here the superscript ∗ denotes the complex conjugate. Let λ=γ+2π i ν and \(\hat u_{n} =R_{n}+i I_{n}\), then y n (t) becomes
and the contribution of excitatory and inhibitory currents to power in a certain frequency ν, in different populations can be defined by Eq. (18).
Rights and permissions
About this article

Cite this article
Hashemi, M., Hutt, A. & Sleigh, J. How the cortico-thalamic feedback affects the EEG power spectrum over frontal and occipital regions during propofol-induced sedation. J Comput Neurosci 39, 155–179 (2015). https://doi.org/10.1007/s10827-015-0569-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10827-015-0569-1