
Abstract
Cell-penetrating peptides (CPPs) are short length permeable proteins have emerged as drugs delivery tool of therapeutic agents including genetic materials and macromolecules into cells. Recently, CPP has become a hotspot avenue for life science research and paved a new way of disease treatment without harmful impact on cell viability due to nontoxic characteristic. Therefore, the correct identification of CPPs will provide hints for medical applications. Considering the shortcomings of traditional experimental CPPs identification, it is urgently needed to design intelligent predictor for accurate identification of CPPs for the large scale uncharacterized sequences. We develop a novel computational method, called TargetCPP, to discriminate CPPs from Non-CPPs with improved accuracy. In TargetCPP, first the peptide sequences are formulated with four distinct encoding methods i.e., composite protein sequence representation, composition transition and distribution, split amino acid composition, and information theory features. These dominant feature vectors were fused and applied intelligent minimum redundancy and maximum relevancy feature selection method to choose an optimal subset of features. Finally, the predictive model is learned through different classification algorithms on the optimized features. Among these classifiers, gradient boost decision tree algorithm achieved excellent performance throughout the experiments. Notably, the TargetCPP tool attained high prediction Accuracy of 93.54% and 88.28% using jackknife and independent test, respectively. Empirical outcomes prove the superiority and potency of proposed bioinformatics method over state-of-the-art methods. It is highly anticipated that the outcomes of this study will provide a strong background for large scale prediction of CPPs and instructive guidance in clinical therapy and medical applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
According to cellular anatomy, cell is the fundamental unit of life [1]. A cell is surrounded by fluid flexible phospholipid bilayer called plasma membrane [2], that controls the passage of biomolecules and certain small-sized materials via active and passive transport [3]. Many potential therapeutic drug targets [4] are located within the interior of the cell that requires the entry of pharmaceutical molecules for effective treatment. However, these biomolecules couldn't pass membrane which hampers their biological functions. To deliver diagnostic tiny genetic materials within the cell nucleus via traditional techniques is formidable and challenging [5] because these clinical methods e.g., (electroporation and microinjection) have a severe impact on the human body and impair the biological activities of cell [6, 7]. Fortunately, about three decades ago, the discovery of CPPs have opened a promising perspective to treat various devastating diseases such as cancer treatment, Gene therapy, [8] and acquired immunodeficiency syndrome (AIDs) [9, 10]. CPPs are relatively shorter proteins nearly (30–50) amino acid residues, have become an influential carrier of drugs, genes, proteins, and nanoparticles into a cell without significant loss of membrane integrity [11,12,13]. CPPs are also called cell-permeable proteins [14], act as a transmembrane vector to penetrate diverse physiological barriers such as blood–brain barrier [15], skin dermis, and gastroenteric mucosa [16]. The high translocation is a unique property of CPPs because of negligible toxicity and cationic nature [17]. In addition to the advantages of peptides drugs as a safe carrier of various therapeutic agents inside the cell [18,19,20], CPPs have been applied successfully in both in-vitro and in-vivo [21], under the normal physiological conditions [22, 23]. Furthermore, CPPs are favorable with other drug vectors to develop a novel multifunctional cargo-carrying platform that accelerates the targeted delivery of drugs and stability during blood circulation [24, 25]. In addition, compared with traditional chemotherapy, peptide-based therapy has numerous benefits, likewise low production price, high specificity and ease of synthesis [26]. Thus, to keep in view the medical applicability as a potent intracellular delivery candidate to treat various therapeutic diseases [27, 28], CPPs composition knowledge is still under development. In this regard, it is very crucial for biomedical drug development and basic life science research to discriminate CPPs from peptides either natural or designed artificially. Unluckily, wet-laboratory based methods [29, 30] of CCPs identification are overpriced, enormously slow and resource intensive. Furthermore, the high error rate of false-positive and false-negative hamper a clear image of the intercome at large. Therefore, due to outburst of sequences in world bank databases triggerd the demand for a novel intelligent automated method.
During the preceding decades, many computational approaches have been proposed to predict cell penetrating peptides solely from their primary sequence (see, e.g. [31,32,33,34,35,36,37].). Hansen et al. [31], designed the pioneer bioinformatics model for identifying CPPs using z-scales of the physiochemical descriptor. The model was trained on 87 non-redundant CPPs. Dobchev et al. [32], proposed another computational system for CPPs classification, using an artificial neural network (ANN) in conjunction with biochemical features of quantitative structure–activity relationship (QSAR) on 101 peptides. They improved the prediction accuracy up to (80%-100%) using principle component analysis (PCA) as a dimension reduction technique. Similarly, Sanders et al. [33], constructed the state-of-the-art sequence-based cell penetrating peptides predictor on a more objective benchmark dataset, which utilizes 61 important physicochemical attributes embedded into support vector machine (SVM) learning engine for classification. The overall accuracy, sensitivity, and specificity were 75.86%, 75.90%, and 76.80%, respectively. Subsequently, Gautam et al. [34], released CPPsite for true CPPs prediction using different non-sequential encoding schemes. For example, simple amino acid composition (AAC), binary pattern profile and dipeptide composition (DPC) in conjunction with SVM algorithm. However, the drawback of using DPC feature cause sparsity due to short peptide sequence. Furthermore, to increase the generalization power of prediction model, Holton et al. [35], constructed a web-based predictor, called CPPpred, which employed N-to-1 Neural Network. Chen et al. [36], adopted random forest as a training model with a combination of pseudo amino acid composition (PseAAC) attributes for discriminating CPPs from non-CPPs. However, they did not implement a web server, which bounds the applicability of the proposed method. Tang et al. [37], developed an online CPP predictor, called C2Pred, based on the SVM algorithm in fivefold cross-validation to identify CPPs. In this model, analysis of variance (ANOVA) was employed to select optimal attributes extracted from g-gap dipeptide composition. In their study, they failed to consider robust feature representation methods that describe the physiochemical properties of peptides. Diener et al. [38] improved the prediction performance by using the frequency of conventional amino acid composition and biochemical features. Similarly, Wei et al. [39] developed a public web server, called SkipCPP-Pred by exploiting distance information of peptide sequence using adoptive k-skip-n-gram feature method with random forest algorithm for Accurate classification of CPPs. More recently, the same author Wei et al. [40] developed a sequential bioinformatics method called, CCPred-RF for recognizing CPPs. In this study, the author used conventional methods such as adaptive gap DPC, parallel correlation pseudo amino acid composition (PC-PseAAC), physicochemical properties and fed the optimal features to RF.
Despite the tremendous effort in advancing automated CPP identification, the performance of existing methods is still unsatisfactory, to fill this gap following considerations are essential to be addressed for building a reliable predictor. First, CPPs are short proteins in the range of (5–30 amino acid residues). How to effectively extract the deeply hidden patterns in complicated peptides segments is a major challenge that fully explored the properties of CPPs. Second, in recent research, it has been demonstrated that individual descriptors not enough to capture useful information [41], in such circumstances integrated features capable to extract sufficient patterns and enhance the predictor efficiency [42, 43]. Third, a suitable feature selection strategy is extremely important for removing redundant attributes, overfitting issue, and improving the prediction performance of the system. Fourth, new machine learning algorithms are desired to classify CPPs with high true predictions. Thus, highlighting the aforementioned deficiencies, we develop an intelligent sequence-based system named, TargetCPP, for classifying cell-penetrating peptides.
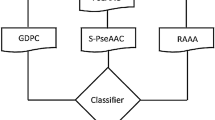
The proposed TargetCPP predictor entails the following main steps to accomplish this task. For a given protein sample, TargetCPP captures four types of key features i.e., composite protein sequence representation (CPSR) [44], composition transition and distribution (CTD), split amino acid composition (SAAC) [45] and information theory features (ITF). Based on multi-perspective patterns the features were fused in different combination and performed experiments to generate novel features. Next, we employ a powerful mRmR feature selection algorithm to select salient feature vectors as a final subset of each peptide. Finally, we utilize gradient boost learning engine to build the prediction model for targeting CPPs. We quantitatively assessed the efficacy of the proposed TargetCPP method, both on the training dataset and independent testing and proves that our model outperformed existing state-of-the-art approaches. The workflow of TargetCPP predictor is illustrated in Fig. 1.

Schematic workflow of the proposed TargetCCP method
Methods and materials
Data collection
The fundamental step of designing a promising predictor is to establish a high-quality stringent benchmark dataset [44]. In this work, we have utilized two datasets derived from [33, 39], to train and test the proposed method for CPPs identification. The well-known CD-HIT program [46], is used to avoid homology bias and redundancy with ≤ 80% for any two peptide samples. Thus, after the above screening method, finally, 462 non-redundant true CPPs and an equal number of non-CPPs were collected with the name CPP924 that is online available on (https://server.malab.cn/SkipCPP-Pred/Index.html) [39]. An independent validation test justifies the generalization power of the developed approach [47]. The final independent dataset after applying homology reduction method [33] with 90% similarity comprised of 111 known CPPs and 34 confirmed non-CPPs sequences. The statistics of both training and independent datasets illustrated in Table 1.
Feature extraction schemes
The prime consideration, how to formulate biological primary peptides into numerical values or extract informative patterns for identifying CPPs is a quite challenging and fascinating task worthy of study in developing a statistical prediction [45]. Numerous feature extraction schemes have been developed, such as AAC [48], DPC [49] and tripeptide composition (TPC) [50], etc., however, these common conventional methods fail to work due to sequence lose information. In this regard to capture novel pattern characteristics and correlation information among residues, we respectively present new feature representation strategies i.e., composite protein sequence representation (CPSR), composition transition and distribution (CTD), split amino acid composition (SAAC), and information theory features (ITF). The input feature schemes are discussed in detailed as follows.
Composite protein sequence representation (CPSR)
A peptide is a polymer of proteins sequences which contain twenty natural amino acids residues. All residues are structurally same however, possesses unique physiochemical characteristics due to a functional group [51]. These biochemical patterns play a vital role in different protein function prediction problems [52, 53]. CPSR method is implemented to describe the important information of CPPs sequence. CPSR-derived method has been broadly adopted by [44, 54] regarding the chemical attributes of a protein sequence.
Table 2 illustrates the total generated feature space i.e., (75-D) which is distributed in eleven sets with the corresponding number of features in each set.
- Amino acid composition (AAC):
-
In feature encoding methods AAC is the simplest formulation technique representing the peptide samples. This mechanism considers the normalize occurrence frequency of twenty amino acids in a peptide sample [55]. In AAC composition, the peptide can be expressed in the 20-D vector [56].
- Sequence Length L:
-
The total number of native amino acids in the given peptide samples is defined by L [54].
- 2-G exchange group frequency:
-
Bi-Gram exchange group composition plays a significant role to represent the feature set from the primary peptide sample. The exchange groups consider general categories of amino acid residues which make their clusters based on the evolution effects. As a result, thirty-six attributes 36-D are generated from its equivalent 6-letter exchange group of amino acids by computing the frequency of each possible bi-gram pair [51] as details given in supplementary file Table S1.
- Hydrophobic group:
-
Hydrophobicity is the physicochemical property of amino acid denoting the tendency of the water-loving molecule [57] as shown in Table 2. Thus, a peptide sequence in the hydrophobic group is represented by 2-D feature space.
- Electronic group:
-
Table 2 illustrates, amino acid molecules classified into six categories, i.e., electron donor or Acceptor, weak electron donor or Acceptor, electrically neutral, and special amino acid. The six 6-D corresponding attributes are extracted by replacing the amino acid in the peptide chain into its equivalent electron group representation.
- Sum of hydrophobicity:
-
In Table S2 of the supplementary file, the Eisenberg physicochemical index values for 20 amino acid [58] are tabulated which is implemented as a feature set in this study. The hydrophobic index is often used to measure the hydrophobic affinity associated with each amino acid molecule [59]. The normalized index values for least hydrophobic R and most hydrophobic are in the range between − 2.53 and 1.38, respectively.
- Sum of hydrophilicity:
-
Hydro means “water” and philic means “soluble”. Thus, the tendency of proteins that are water-soluble or affinity in water, called hydrophilicity. This is proteins important feature help in recognizing their function.
- Sum of rigidity:
-
In daily life, rigidity has many meanings but its concept originally derived from mechanical engineering that describes the static properties of protein structure under the external influence [60].
- Sum of flexibility:
-
The flexibility of peptide occurs universally at the level of amino acid side-chains and crucial for catalysis and binding function [61].
- Sum of irreplaceability:
-
From a nutritional perspective, nonessential residues are irreplaceable and vital for the physiological process of organisms. The irreplaceability is a response to mutation deterioration in the course of the evolution of life [58]. To compute the irreplaceability of amino acid residues, averaged mutation deterioration (AMD) can be used [58].
- R-group:
-
In peptide, each residue has an identical shape but chemically diverse side chain. However, some of the properties contain the same functional groups [54]. Consequently, five sub-families are made by clustering [62] as given in supplementary Table S1.
Composition Transition and Distribution (CTD)
CTD is global distribution pattern of proteins physicochemical features, initially proposed by Dubchak et al. [63] for protein folding class prediction. CTD has been proven to be successfully used in various prediction problems including prediction of antimicrobial peptides [64], subcellular localization, and protein function classification [65, 66]. Encouraged by this, we implemented CTD feature descriptor strategy to extract seven amino acid properties hydrophobicity, polarizability, solvent Accessibility normalized van der Waals volume, polarity and predicted secondary structure [67]. In this encoding strategy, the C index characterizes the percent composition of each group in primary peptide/protein; the T index denotes the transition likelihood between two adjacent residues of proteins associated with different families [67]. Meanwhile, the index D represents the distribution of amino acid along the sequence of each group in percent (25%, 50%, and 75% or 100% residue respectively) [68]. In the present work, CTD generated a 147-D vector, against each peptide sequence, for detail description and formulation of this method reader are referred to [69].
Information Theory Feature (ITF)
Shannon Entropy
Shannon entropy has been successfully used in PTMs identifications [70]. It computes amino acid preservation in sequences to generate a 1-D feature vector for each residue [71]. For a given query of a primary peptide, the following formulation is used to compute the Shannon entropy.
where Pi denotes the probability of twenty amino acids i in the peptide sequence.
Relative Shannon Entropy
“Relative Shannon Entropy measures the conservation of amino acids compared with the background distribution” [71]. It can be mathematically denoted as:
where Pj = 1/L is the uniform likelihood of the entire sequence of length L. RE is always non-negative and become zero when Pi = P0. Relative entropy generates 1-D feature vector like entropy for each peptide sequence [72].
Information gain score
Information gain score calculates the transformation of information in a sequence fragment influenced by a grouping factor [71]. It can be computed by subtracting RE from has the following formula:
Split amino acid composition (SAAC)
Proteins are a polymer of twenty different amino acid residues. It has been observed that traditional methods cannot dig the crucial information from proteins specifically at their C- and N terminus. SAAC [45] is a novel feature encoding technique capable of extracting the hidden complementary information concealed in fragments. SAAC based approach has been widely adopted in many problems of protein predictions [73,74,75]. In this method, the given query peptide sequence is segmented into N- and C terminus and the composition of each part is calculated distinctly [76]. In our study, According to the sequence length, we divided the peptides into three segments: (i) 5 amino- acid of C termini, (ii) 5 amino acid of N termini, and (iii) region between these two termini. Unlike conventional AAC the generated feature dimension is 60-D instead of 20-D. It can be mathematically formulated as:
where C- and N terminus are represented by C and N alphabets respectively.
Feature Selection
Feature selection methods have become an apparent prerequisite in numerous pattern recognition and bioinformatics applications [77]. Because the existence of redundant information and noise influence of different features may potentially cause over or underfitting [78], which seriously deteriorate the generalization capability of the prediction model. In this regard, we implement two steps feature selection technique to choose optimal attributes, reduce time complexity and enhance model performance [79]. In the first step, we determined the F-Score to analyze feature importance that can provide a better insight into the biological significance of features characteristics [80]. In the second step, we employ a powerful feature selection algorithm called minimum redundancy and minimum relevancy (mRmR) [81]. In recent years, mRmR algorithm has been applied to various biological problems [82,83,84]. It is a widespread multivariate feature selection technique, was originally introduced by Peng et al. [81]. This heuristic strategy evaluates the investigated attributes for a certain problem by providing two lists, maximum relevance (MaxRel) attribute list and minimum redundancy (MinRed) attribute list [85]. MaxRel attribute list implies that removing the attributes with the lowest relevance to the target variable, while Min-Red means that removing the attributes with the maximum redundancy to the attributes already selected.
Firstly, the mutual relationship between the attributes and the class labels can be computed by mutual information (MI), because MI is the measuring tendency of two variables relevance and redundancy in mRmR [86]. Let’s say mi and y are two vectors, then redundancy and relevancy between vectors (attributes and target class) can be calculated by following MI mathematical expression [87].
Here the above equation is probability density function where P (mi), P(y) represents marginal probabilistic densities and P (mi, y) denotes the joint probabilistic density.
Consequently, the maximum relevance can be formulated as:
In Eq. (6), max \(({\text{Re}} l)\) denotes maximum relevancy between two vectors i.e. mi features in S sample space and y target. Minimum redundancy can be formulated as:
In Eq. (7), min \(({\text{Re}} d)\) denotes minimum redundancy between mi and mj.
Eventually, by concatenating Eqs. (6) and (7), we get
Thus, after employing mRmR method on the entire original feature vector S, a ranked feature vector SmRmR is obtained as denoted by the following expression:
where high ranked (low index) features have good prediction capability and low ranked (high index) features have less prediction quality. In this article, we adopted the sequential forward selection (SFS) search method in order to gain the optimal subset. The working principle of SFS strategy is that it eliminates the lowest ranked features in descending order from feature space one by one and transform the training dataset repeatedly to rebuild the final prediction model through leave-one-out cross-validation test. This process continues until the best predictions are obtained by reduced optimal features. The F-score of ranked features in descending order are provided in supplementary file S3. For further detail (see “Results and discussion” section).
Classification algorithms
In decision-making system classification is a well-known type of supervised learning. In our study, we use three different classifiers: Gradient boost decision tree (GBDT) [88], Decision Tree (DT) [89], and Naïve Bayes (NB) [90] for predicting CPPs and Non-CPPs peptides. However, due to outstanding learning performance compare the other two algorithms we use GBDT to build the prediction model.
Gradient boost decision tree classifier (GBDT)
Over the past years, GBDT learning algorithm [91] has been widely used by the researchers from a broad spectrum in computational biology and bioinformatics applications [92, 93]. Unlike other ensemble approaches, for example, Random forest and AdaBoost, GBDT construct a scalable and accurate base predictive model from a non-linear ensemble of weak learners, typically decision trees [94]. In GBDT, the learning procedure fits new models in order to facilitate a more accurate estimate of the response variable [95].”The basic concept of this classifier is to design the new base-learners to be maximally correlated with the negative gradient of the loss function, related to the entire ensemble” [94].
Assumed that there are N number of training samples:\(\left\{ {\left( {a_{1} ,b_{1} } \right).....\left( {A_{N} ,B_{N} } \right)} \right\}\), where.
Ai ∈ A ⊂ Rn, bi ∈ B ⊂ R. The GBDT classifier estimates the functionality of predicted peptides a by linear concatenation of single decision trees, see Eq. 10.
where \(T\left( {a;\theta_{k} } \right)\) the i-th decision tree is θk is its parameter and K represent the number of decision trees.
The GBDT classifier computes the final estimation in a forward stage-wise fashion. Let’s imagine fo (a) is the initial model of a, the model in m step can be obtained by Eq. 11.
where \(f_{k - 1} \left( a \right)\) is the model in k-1 steps. The risk minimization parameter \(\theta_{k}\) is formulated in Eq. 11.
where the loss function is denoted by L in Eq. 12.
The main objective of assuming the base function linear additivity is to estimate the \(\theta_{k}\) for best fitting the residual \(L\left( {b - f_{k - 1} \left( a \right)} \right)\). At the end, the negative gradient of the loss function \(f_{k - 1}\) is used to estimate the residual approximation.
where i is the index of i-th sample. Finally, we train a decision tree model by all \(R_{ki} ,i \in \left[ {1, \ldots ,N} \right]\) for estimating the parameter \(\theta_{k}\).
The decision tree parameter is employed to split input variables space into a uniform frame area by tree-based rule system. Each tree partition maps to an if–then rule over some input variables. This structure of the decision tree naturally models the interactions between predictor variables. If the parameter maps the input space A into J disjoint regions R1…, RJ, and the output is cj for each region Rj, then the Tree can be written as in Eq. 14.
The concept of GBDT classifier is represented as pseudo-code in Algorithm 1 below.

Model assessment and cross-validation
We evaluate the performance of TargetCPP model using six classification measures including Accuracy (ACC), Specificity (Spe), Sensitivity (Sen), G-mean and F-measure and Mathews’ Correlation coefficient (MCC) respectively. The ACC compute the true predictions of the model. The Spe computes the percentage of negative samples that are predicted correctly. The Sen describes the true positive rate of the considerd class. The G-mean statistical index is employed for the predictor balance performance between negative and positive class. The F-measure corresponds to the harmonic mean of precision and recall. The MCC measure is needed both for under and over-prediction in binary classification. This measure takes the values in the range of [− 1 1]. A value of -1 denotes disagreement and vise versa. These metrics are always counted on the basis of four different values which includes true positive (TP), true negative (TN), false positive (FP) and false negative (FN). Mathematically, they are defined as;
where
and
In the above Eqs. (15–22), TP represents correctly recognized CPPs and TN denote correctly recognized non-CPPs whereas FP denotes non-CPPs that are mistakenly predicted as the CPPs and FN represent CPPs that are predicted as non-CPPs, respectively.
In designing an automated model, the following three evaluation methods are commonly used to analyze the quality of the predictor: K-fold or Sub-sampling test, leave-one-out or Jackknife test and independent test [96]. Among these cross-validation tests, rigorous jackknife method is deemed more effective and stringent for examine the performance. In the jackknife cross-validation, we employ each sample of the benchmark dataset as test dataset one by one, and the remaining are used to train the model [97]. In this study, both jackknife and independent cross-validation are adapted to judge the effectiveness and generalization power of our constructed model.
Results and discussion
In this section, we elucidate the detailed empirical outcomes of TargetCCP predictor for correctly targeting CPPs. The proposed method capture four types of key features i.e., CPSR, CTD, SAAC and ITF from peptide sequence. Based on multi-perspective patterns the features were fused in different serial combination and performed experiments to generate novel features. Next, we employ a suitable mRmR feature selection algorithm to select rich attributes as a final subset from the whole feature set. Finally, we then utilize gradient boost decision tree classifier to build the final prediction model for recognizing CPPs. We quantitatively assessed the efficacy of the proposed TargetCPP method, both on jackknife and independent testing and proves that our model outperformed the advanced recent approaches in the literature. We have elaborated our experiments in subsequent section as follows:
Performance analysis of individual features
In this subsection, we examine the predictive performance of single descriptors, i.e., CPSR, CTD, SAAC, and ITF using various classifiers i.e., DT, NB, and GBDT. Each feature method is evaluated by performing jacking test on the benchmark training dataset CPP924 with six performance metrics of binary classification i.e., ACC, Sen, Spe, MCC, F-measure, and G-Mean. Table 3 summarizes the success rates comparison of individual feature space using three learning hypotheses.
The ACC of CPSR using DT, NB, and GBDT algorithms are 83.09%, 82.48%, and 84.91%, and MCC values are 0.671, 0.649, and 0.698 points. The second best prediction in term of ACC and MCC are obtained by SAAC feature space using GBDT classifier which is 83.21% and 0.664 points, respectively. Likewise, the prediction performance of ITF features using the same set of classifiers achieved satisfactory results which are ACC = 81.24%, 80.63%, and 81.72% and MCC = 0.624, 0.629 and 0.642 points for DT, NB and GBDT algorithms. CTD feature descriptor generated worse results by GBDT algorithm i.e., ACC of 78.46% and MCC of 0.576 points. From Table 3, we can easily observe that CPSR generally produce good recognition for all the mentioned algorithms compare, the other three individual sample space concerning the six evaluation measures. The underlying reason for CPSR performing better prediction comparing the remaining three feature descriptor is that CPSR consist of new biochemical information (rigidity, flexibility, and irreplaceability) that contribute directly related patterns for targeting CPPs.
Performance analysis of ensemble features
In the current subsection, we further enhance the performance of CCP predictor by integrating four promising features spaces in various combinations. We empirically investigate to determine which feature combination can better contribute to the prediction of CPPs. In this regard, we performed a series of different comparative analysis. Table 4 reports the prediction outcomes of six different combinations using three classification algorithms by jackknife test on CPP924. In the case of hybrid features, we achieved progressive results when all features were fused. The highest performance achieved by GBDT classifier nearly reached to 90.68% ACC, 90.90% Sen, 90.47% Spe, and 0.814 points MCC. In contrast, the poorer performance is achieved by SAAC + CTD feature view using DT classifier which is ACC = 76.15%, Sen = 72.26%, Spe = 80.04%, and MCC = 0.524 points. The significant improvement in discrimination of CCPs from non-CPPs in Table 4 raises an interesting question: Can ensemble features positively impact in building a predicting model to target correct CPPs? The answer to this question is yes because single-view features some time fails to capture the valuable information from peptide samples, which might cause a poor predictive outcome. In such circumstances, hybrid features can better represent CCP hidden information which positively influences the performance of intelligent TargetCPP predictor.
Performance analysis after feature selection
It is obvious that in the previous section the prediction performance of CPP identification was improved by hybridizing four features i.e., CTD, CPSR, SAAC, and ITF in distinct combinations. In total, the dimension of these nominal feature space is 285-D including 147 CTD properties, 75 CPSR properties, 60 SAAC properties, and 3 ITF properties. However, to overcome the risk of overfitting during model training and further boost-up the generalization power of the proposed TargetCPP method, a suitable features selection method is quite significant. In the present research, we apply a powerful two-step mRmR feature selection algorithm on the integrated attributes and select 80 top-ranked features based on minimum redundancy and close relevancy rule to learn the final CPP model. Concretely, we appraise the discrimination power of classifiers on the optimized features subset using the jackknife cross-validation method. Table 5 demonstrates the success rate comparison of supervised learning algorithms before and after feature selection on the training dataset CPP924. It can be seen from Table 5 that the GBDT and NB classifiers have improved the performance in term of all six evaluation measures while DT classifier performance has degraded after feature selection. The average ACC of GBDT increased from 90.48% to 93.54% and MCC value jumped from 0.814 to 0.871 points which are 3.07% and 0.62 points higher than before reduced features. Similarly, the prediction performance of NB in term of ACC and MCC enhanced from 86.05% to 90.14% and from 0.737 to 0.803 points respectively. However, the ACC and MCC of DT classifier decreased by 1.39% and 0.027 points. Thus, it can be seen from Fig. 2a, b that GBDT produced consistent results and DT, in contrast, generated worst results before and after using mRmR algorithm. Similarly, the visual influence of utilizing mRmR method on the feature vectors is depicted by t-distributed Stochastic Neighbor Embedding (t-SNE) in Fig. 3a, b. To analyze the contribution of best feature vectors from the four types of feature spaces are in the ratio: 0.129(19/147) features are selected from CTD method, 0.493(37/75) are selected from CPSR method, 0.366 (22) are selected from SAAC method, and 0.66 (2/3) ITF features are selected. Thus, the aforementioned calculation demonstrates that all the features particularly ITF and CPSR incorporate in targeting the correct CPP predictions.

a Receiver Operating Characteristic (ROC) curves of classifiers before feature selection over jackknife cross-validation test on the training dataset. b Receiver Operating Characteristic (ROC) curves of classifiers after feature selection over jackknife cross-validation test on the training dataset

a 2-D t-SNE scatters plot of CPPs and Non-CPPs before feature selection on the training dataset. The number 0 in the legend with gray color represents the CCP class and the number 1 with yellow color represent Non-CPPs class. b 2-D t-SNE scatters plot of CPPs and Non-CPPs after feature selection on the training dataset. The number 0 in the legend with gray color represents the CCP class and the number 1 with yellow color represent Non-CPPs class
Comparison of TargetCPP with existing predictors on training dataset CPP924
In this subsection, we compare the predictive capability and effectiveness of the TargetCPP with recently published methods including CellPPD-DC, CellPPD-BP, SkipCPP-Pred [39], Diener’s method [38], and CPPred-RF [40] on the benchmark dataset CPP924. Table 6 depicts the performance outcomes of previous methods derived from Wei et al. CPPred-RF research [40]. For a fair comparison, we draw a bar graph representation along with four performance measures in Fig. 4a. Our developed forecasting system TargetCPP substantially outperformed all the predictors for CPP identification in term of four evaluation metrics such as ACC, Sen, Spe, and MCC. Our method achieved Accuracy of 93.55%, Sen of 92.15% which means truly predicted CPPs, 94.90% Spe which means truly predicted Non-CPPs and 0.873 points MCC, respectively. It is worth noting that Wei L et al. [39] constructed three predictors shortly named as CellPPD-DC, CellPPD-BP, and SkipCPP-Pred which achieved ACC of 87.00%, 83.70%, and 90.60%, as well as the values of MCC, are 0.745, 0.680 and 0.812 points. To be specific, the method of Diener’s et al. [38] have a 2.35% decrease in ACC and 0.48 points decrease in MCC as compare to our proposed method. The CPPred-RF [40] is runner-up which produced less 1.90% ACC, 1.69% Sen, 1.40% Spe, and 0.42 points MCC, respectively. Thus, according to reported results, our developed model performed efficiently than the other predictors. To more intuitively summarize the higher discriminative capability of TargetCPP is reflected by several reasons: First, we captured the key diverse information of true CPPs and non-CPPs by incorporating a multi-perspective feature representation strategy. Second, optimal feature selection method enabled our predictive model with fewer features (80) than previous best CPPred-RF predictor (290), this means that our model is faster and helpful for a large amount of CPP predictions.

a Performance comparison of TargetCCP method with existing CPP predictors on benchmark dataset CPP924. b Performance comparison of TargetCCP method with existing CPP predictors on independent dataset Sind
Comparison of TargetCPP with existing predictors on independent dataset CPPind
In order to examine the generalization power of the proposed trained model, we performed independent validation test and compare the outcomes with published methods of Sender et al. [32], Chen et al. method [36] and C2Pred [37] over the four performance measures ACC, Sen, Spe, and MCC. It is clear from Table 7 that TargeCPP obtained the best prediction of ACC 88.27% and MCC 0.771 points which is about 12.41% ACC higher from Sender et al. method [32]. To be specific, the runner-up method C2Pred [37] achieved 83.60% overall ACC while MCC values are not available (N/A). Thus, owing to the best performance of TargetCPP bioinformatics tool, it is anticipated that our work is promising and superior for identification of CPPs (Table 7).
Conclusion
Cell-penetrating peptides are the most important class of peptides, capable of delivering drugs safely inside live cells. In-depth knowledge about CPPs is conducive to design therapeutics drugs for disease treatment. Thus, we developed a new sequence-based CCP predictor called TargetCPP. The empirical outcomes anticipate that our proposed novel TargetCPP holds high potential compared to available advanced CPP methods. The excellent throughput of TargetCPP is due to several reasons including proper feature engineering schemes, intelligent feature optimization algorithm and careful designing of the learning model. The source code, training and independent datasets used in this research has provided in Supplimentary file S4. For convenience, in the future, a publically accessible web server will be established for the proposed method to facilitate peptide-based therapy in particular and life science research in general.
References
Chou K-C, Shen H-B (2007) Recent progress in protein subcellular location prediction. Anal Biochem 370(1):1–16
Krogh A et al (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580
Lennernäs H et al (1996) Comparison between active and passive drug transport in human intestinal epithelial (Caco-2) cells in vitro and human jejunum in vivo. Int J Pharm 127(1):103–107
Ramsey JD, Flynn NH (2015) Cell-penetrating peptides transport therapeutics into cells. Pharmacol Ther 154:78–86
Khalili P et al (2006) A non–RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol Cancer Ther 5(9):2271–2280
Trabulo S et al (2013) Cell-penetrating peptides as nucleic acid delivery systems: from biophysics to biological applications. Curr Pharm Des 19(16):2895–2923
Gebbing M et al (2015) Gene therapeutic approaches to inhibit hepatitis B virus replication. World J Hepatol 7(2):150
Michiue H et al (2014) The acceleration of boron neutron capture therapy using multi-linked mercaptoundecahydrododecaborate (BSH) fused cell-penetrating peptide. Biomaterials 35(10):3396–3405
Frankel AD, Pabo CO (1988) Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55(6):1189–1193
Green M, Loewenstein PM (1988) Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 55(6):1179–1188
Fonseca SB, Pereira MP, Kelley SO (2009) Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev 61(11):953–964
Nakase I et al (2011) Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc Chem Res 45(7):1132–1139
Lakshmanan M et al (2012) Rapid and efficient gene delivery into plant cells using designed peptide carriers. Biomacromol 14(1):10–16
Rüter C et al (2010) A newly identified bacterial cell-penetrating peptide that reduces the transcription of pro-inflammatory cytokines. J Cell Sci 123(13):2190–2198
Zhang D, Wang J, Xu D (2016) Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J Control Release 229:130–139
Patel A, Cholkar K, Mitra AK (2014) Recent developments in protein and peptide parenteral delivery approaches. Therapeutic delivery 5(3):337–365
Soler M et al (2014) Identification of BP16 as a non-toxic cell-penetrating peptide with highly efficient drug delivery properties. Org Biomol Chem 12(10):1652–1663
Kato T et al (2016) Plasmid DNA delivery by arginine-rich cell-penetrating peptides containing unnatural amino acids. Bioorg Med Chem 24(12):2681–2687
Farkhani SM et al (2016) Enhanced cellular internalization of CdTe quantum dots mediated by arginine-and tryptophan-rich cell-penetrating peptides as efficient carriers. Artificial cells, nanomedicine, and biotechnology 44(6):1424–1428
Mäger I et al (2012) The role of endocytosis on the uptake kinetics of luciferin-conjugated cell-penetrating peptides. Biochim Biophys Acta (BBA) 1818(3):502–511
Zaro JL, Shen W-C (2015) Cationic and amphipathic cell-penetrating peptides (CPPs): Their structures and in vivo studies in drug delivery. Front Chem Sci Eng 9(4):407–427
Zhang R et al (2006) Molecular modeling studies of peptide drug candidates against SARS. Med Chem 2(3):309–314
Chou K-C et al (2006) Progress in computational approach to drug development against SARS. Curr Med Chem 13(27):3263–3270
Zhang Q, Gao H, He Q (2015) Taming cell penetrating peptides: never too old to teach old dogs new tricks. Mol Pharm 12(9):3105–3118
Yang Y et al (2015) Synergistic targeted delivery of payload into cancer cells using liposomes co-modified with photolabile-caged cell-penetrating peptides and targeting ligands. J Control Release 213:e128
Otvos L (2008) In Peptide-based drug design. Peptide-based drug design: here and now, Springer. pp 1–8.
Raucher D, Ryu JS (2015) Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med 21(9):560–570
Simon MJ et al. (2010) Evaluation of the cell-penetrating peptide TAT as a trans-blood-brain barrier delivery vehicle. In: Bioengineering Conference, Proceedings of the 2010 IEEE 36th Annual Northeast. IEEE
Gao S et al (2011) An unusual cell penetrating peptide identified using a plasmid display-based functional selection platform. ACS Chem Biol 6(5):484–491
Lee JH et al (2012) Screening of cell-penetrating peptides using mRNA display. Biotechnol J 7(3):387–396
Hansen M, Kilk K, Langel Ü (2008) Predicting cell-penetrating peptides. Adv Drug Deliv Rev 60(4–5):572–579
Dobchev A et al (2010) Prediction of cell-penetrating peptides using artificial neural networks. Curr Comput-Aided Drug Des 6(2):79–89
Sanders WS et al (2011) Prediction of cell penetrating peptides by support vector machines. PLoS Comput Biol 7(7):e1002101
Gautam, A., et al., CPPsite: a curated database of cell penetrating peptides. Database, 2012. 2012.
Holton TA et al (2013) CPPpred: prediction of cell penetrating peptides. Bioinformatics 29(23):3094–3096
Chen L et al (2015) Prediction and analysis of cell-penetrating peptides using pseudo-amino acid composition and random forest models. Amino Acids 47(7):1485–1493
Tang H et al (2016) Prediction of cell-penetrating peptides with feature selection techniques. Biochem Biophys Res Commun 477(1):150–154
Diener C et al (2016) Effective design of multifunctional peptides by combining compatible functions. PLoS Comput Biol 12(4):e1004786
Wei L, Tang J, Zou Q (2017) SkipCPP-Pred: an improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genomics 18(7):1
Wei L et al (2017) CPPred-RF: a sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J Proteome Res 16(5):2044–2053
Zhang L et al (2015) An ensemble method to distinguish bacteriophage virion from non-virion proteins based on protein sequence characteristics. Int J Mol Sci 16(9):21734–21758
Zhang Y-N et al (2012) Predicting protein-ATP binding sites from primary sequence through fusing bi-profile sampling of multi-view features. BMC Bioinform 13(1):118
Han GS et al (2013) An ensemble method for predicting subnuclear localizations from primary protein structures. PLoS ONE 8(2):e57225
Kabir M et al (2018) Intelligent computational method for discrimination of anticancer peptides by incorporating sequential and evolutionary profiles information. Chemom Intell Lab Syst 182:158–165
Ali F et al (2018) DBPPred-PDSD: Machine learning approach for prediction of DNA-binding proteins using Discrete Wavelet Transform and optimized integrated features space. Chemom Intell Lab Syst 182:21–30
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13):1658–1659
Wei Z-S et al (2015) A cascade random forests algorithm for predicting protein-protein interaction sites. IEEE Trans Nanobiosci 14(7):746–760
Hu J et al. (2019) TargetDBP: accurate DNA-binding protein prediction via sequence-based multi-view feature learning. IEEE/ACM transactions on computational biology and bioinformatics
Ahmad K, Waris M, Hayat M (2016) Prediction of protein submitochondrial locations by incorporating dipeptide composition into Chou’s general pseudo amino acid composition. J Membr Biol 249:1–12
Kurgan L et al (2009) CRYSTALP2: sequence-based protein crystallization propensity prediction. BMC Struct Biol 9(1):50
Golmohammadi SK et al. (2007) Classification of cell membrane proteins. in Frontiers in the Convergence of Bioscience and Information Technologies, 2007. FBIT 2007. IEEE
Xia X, Li W-H (1998) What amino acid properties affect protein evolution? J Mol Evol 47(5):557–564
Qiu W-R et al (2017) iKcr-PseEns: identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics 110:239–246
Hayat M, Khan A (2011) Predicting membrane protein types by fusing composite protein sequence features into pseudo amino acid composition. J Theor Biol 271(1):10–17
Chen W et al (2016) iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 7(13):16895–16909
Hayat M, Khan A, Yeasin M (2012) Prediction of membrane proteins using split amino acid and ensemble classification. Amino Acids 42(6):2447–2460
Singh O, Su EC-Y (2016) Prediction of HIV-1 protease cleavage site using a combination of sequence, structural, and physicochemical features. BMC Bioinform 17(17):478
Tang H, Chen W, Lin H (2016) Identification of immunoglobulins using Chou's pseudo amino acid composition with feature selection technique. Mol BioSyst 12(4):1269–1275
Eisenberg D et al (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179(1):125–142
Karshikoff A, Nilsson L, Ladenstein R (2015) Rigidity versus flexibility: the dilemma of understanding protein thermal stability. FEBS J 282(20):3899–3917
Huber R (1987) Flexibility and rigidity, requirements for the function of proteins and protein pigment complexes. Portland Press Limited, London
Kedarisetti KD, Kurgan L, Dick S (2006) Classifier ensembles for protein structural class prediction with varying homology. Biochem Biophys Res Commun 348(3):981–988
Dubchak I et al (1995) Prediction of protein folding class using global description of amino acid sequence. Proc Natl Acad Sci 92(19):8700–8704
Bhadra P et al (2018) AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci Rep 8(1):1697
Nair R, Rost B (2005) Mimicking cellular sorting improves prediction of subcellular localization. J Mol Biol 348(1):85–100
Cai C et al (2003) SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res 31(13):3692–3697
Jones A. et al. (2015) RF-Phos: random forest-based prediction of phosphorylation sites. in 2015 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE
Zhang X, Liu S (2016) RBPPred: predicting RNA-binding proteins from sequence using SVM. Bioinformatics 33(6):854–862
Govindan G, AS Nair (2011) Composition, transition and distribution (CTD)—a dynamic feature for predictions based on hierarchical structure of cellular sorting. in India Conference (INDICON), 2011 Annual IEEE. IEEE
Capra JA, Singh M (2007) Predicting functionally important residues from sequence conservation. Bioinformatics 23(15):1875–1882
Wei L. et al. (2017) Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Transactions On Computational Biology and Bioinformatics
Ismail HD, Newman RH (2016) RF-Hydroxysite: a random forest based predictor for hydroxylation sites. Mol BioSyst 12(8):2427–2435
Arif M, Hayat M, Jan Z (2018) iMem-2LSAAC: a two-level model for discrimination of membrane proteins and their types by extending the notion of SAAC into Chou's pseudo amino acid composition. J Theor Biol 442:11–21
Khan ZU, Hayat M, Khan MA (2015) Discrimination of acidic and alkaline enzyme using Chou’s pseudo amino acid composition in conjunction with probabilistic neural network model. J Theor Biol 365:197–203
Ahmad S, Kabir M, Hayat M (2015) Identification of Heat Shock Protein families and J-protein types by incorporating Dipeptide Composition into Chou's general PseAAC. Comput Methods Programs Biomed 122(2):165–174
Mirza MT et al (2013) MitProt-Pred: Predicting mitochondrial proteins of Plasmodium falciparum parasite using diverse physiochemical properties and ensemble classification. Comput Biol Med 43(10):1502–1511
Wang S et al (2016) Analysis and identification of aptamer-compound interactions with a maximum relevance minimum redundancy and nearest neighbor algorithm. BioMed Res Int 102:1567–1577
Cheng F et al (2012) Prediction of chemical-protein interactions network with weighted network-based inference method. PLoS ONE 7(7):e41064
Iqbal M, Hayat M (2016) “iSS-Hyb-mRMR”: identification of splicing sites using hybrid space of pseudo trinucleotide and pseudo tetranucleotide composition. Comput Methods Programs Biomed 128:1–11
Win TS et al (2018) PAAP: A web server for predicting antihypertensive activity of peptides. Future Med Chem 10(15):1749–1767
Peng H, Long F, Ding C (2005) Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell 27(8):1226–1238
Zhang Y, Ding C, Li T (2008) Gene selection algorithm by combining reliefF and mRMR. BMC genomics 9(2):S27
Xu Y et al (2014) Identification of thyroid carcinoma related genes with mRMR and shortest path approaches. PLoS ONE 9(4):e94022
Li Z et al (2014) The prediction of methylation states in human DNA sequences based on hexanucleotide composition and feature selection. Anal Methods 6(6):1897–1904
Cai Y et al (2012) Prediction of lysine ubiquitination with mRMR feature selection and analysis. Amino Acids 42(4):1387–1395
Ding H et al (2013) Prediction of Golgi-resident protein types by using feature selection technique. Chemom Intell Lab Syst 124:9–13
Peng H, Long F, Ding C (2005) Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell 8:1226–1238
Chen T, C. Guestrin (2016) Xgboost: A scalable tree boosting system. In: Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining. ACM
Pal M, Mather PM (2003) An assessment of the effectiveness of decision tree methods for land cover classification. Remote Sens Environ 86(4):554–565
Feng PM et al (2013) Naive Bayes classifier with feature selection to identify phage virion proteins. Computat Math Methods Med. https://doi.org/10.1155/2013/530696
Friedman JH (2001) Greedy function approximation: a gradient boosting machine. Ann Stat 29:1189–1232
Chen F et al (2017) Prediction of luciferase inhibitors by the high-performance MIEC-GBDT approach based on interaction energetic patterns. Phys Chem Chem Phys 19(15):10163–10176
Liao Z et al (2016) In silico prediction of gamma-aminobutyric acid type-A receptors using novel machine-learning-based SVM and GBDT approaches. BioMed Res Int. https://doi.org/10.1155/2016/2375268
Zhou C et al (2017) Multi-scale encoding of amino acid sequences for predicting protein interactions using gradient boosting decision tree. PLoS ONE 12(8):e0181426
Rawi R et al (2017) PaRSnIP: sequence-based protein solubility prediction using gradient boosting machine. Bioinformatics 34(7):1092–1098
Arif M et al (2020) Pred-BVP-Unb: fast prediction of bacteriophage Virion proteins using un-biased multi-perspective properties with recursive feature elimination. Genomics 112(2):1565–1574
Wang Y et al (2017) Improved detection of DNA-binding proteins via compression technology on PSSM information. PLoS ONE 12(9):e185587
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 61772273 and 61373062) and Fundamental Research Funds for the Central Universities (No. 30918011104).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Arif, M., Ahmad, S., Ali, F. et al. TargetCPP: accurate prediction of cell-penetrating peptides from optimized multi-scale features using gradient boost decision tree. J Comput Aided Mol Des 34, 841–856 (2020). https://doi.org/10.1007/s10822-020-00307-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-020-00307-z