
Abstract
The P2Y12 receptor (P2Y12R) is an ADP-activated G protein-coupled receptor (GPCR) that is an important target for antithrombotic drugs. Three homology models of P2Y12R were compared, based on different GPCR structural templates: bovine rhodopsin (bRHO), human A2A adenosine receptor (A2AAR), and human C-X-C chemokine receptor type 4 (CXCR4). By criteria of sequence analysis (25.6% identity in transmembrane region), deviation from helicity in the second transmembrane helix (TM2), docked poses of ligands highlighting the role of key residues, accessibility of a conserved disulfide bridge that is reactive toward irreversibly-binding antagonists, and the presence of a shared disulfide bridge between the third extracellular loop (EL3) and the N-terminus, the CXCR4-based model appeared to be the most consistent with known characteristics of P2Y12R. The docked poses of agonist 2MeSADP and charged anthraquinone antagonist PSB-0739 in the binding pocket of P2Y12R-CXC agree with previously published site-directed mutagenesis studies of Arg256 and Lys280. A sulfonate at position 2 of the anthraquinone core created a strong interaction with the Lys174(EL2) side chain. The docking poses of the irreversibly-binding, active metabolite (existing as two diastereoisomers in vivo) of the clinically utilized antagonist Clopidogrel were compared. The free thiol group of the 4S diastereoisomer, but not the 4R isomer, was found in close proximity (~4.7 Å) to the sulfur atom of a disulfide bridge involving Cys175, suggesting greater activity in covalent binding. Therefore, ligand docking to the CXCR4-based model of the P2Y12R predicted poses of both reversibly and irreversibly-binding small molecules, consistent with observed pharmacology and mutagenesis studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The P2Y12 receptor (P2Y12R), a G protein-coupled receptor (GPCR), which is activated by ADP (1, Fig. 1), has a prominent role in the regulation of platelet aggregation and hemostasis [1]. The P2Y12R on the surface of the platelet is the target of various irreversible and reversible antagonists for the treatment of cardiocirculatory diseases [2]. However, the limitations and disadvantages of the currently available drugs create a need for novel P2Y12R-selective antagonists with less variability in in vivo activity and bioavailability, i.e., less dependent on pharmacogenomic factors. The structures of nucleotide agonists (1,2) and structurally diverse, reversible (3–7) and irreversible (8) antagonists used as ligands to study the P2Y12 receptor are shown in Fig. 1 [3]. Known agonists generally contain a phosphate moiety, while both charged and uncharged antagonists have been reported.

Ligands used to study the P2Y12 receptor, including nucleotide agonists (1,2) and both nucleotide-related (4,5) and nonnucleotide (3, 6–8) antagonists. pEC50 values are shown [3]
Recent advances in protein X-ray crystallography applied to GPCRs have provided many different structures of class A GPCRs. However, at present these GPCR structures cover only a limited region of the GPCR superfamily, with only six GPCRs from four different GPCR subfamilies [4–10]. Only recently was any information available on the active conformations of GPCRs [11–13]. The crystal structure of rhodopsin (RHO) [4] was the first GPCR structure to be determined, followed by the structures of the beta1 and beta2 adrenergic receptors (ADRB1, ADRB2) in 2007 [5–7], and, later, the A2A adenosine receptor (A2AAR) [8]. The recent report of the C-X-C chemokine receptor type 4 (CXCR4) [9] confirmed the expectation that each GPCR subfamily has unique structural features that are difficult to predict with the available computational tools. The crystal structure of the D3 dopaminergic receptor (DRD3), recently published [10], falls in the biogenic amine GPCR subfamily along with the adrenergic receptors and, indeed, shows a high 3-dimensional structural similarity with ADRB1 and ADRB2.
More structures of other GPCRs are expected in the near future, but for many GPCRs it could take a few years before the crystals become available. Thus, predictive modeling methods are still needed to gain insight into the three dimensional structures of many GPCRs and for docking of new ligands to already crystallized receptors. Homology modeling of GPCRs and ligand docking has proven useful for selecting likely interaction sites for mutagenesis studies and as a guide in the rational modification of ligands [14–16]. However, the generation of accurate GPCR models and the design of novel ligands by means of computer-aided techniques are still very challenging for all GPCRs, and particularly for those GPCRs that share a low sequence identity with the available structural templates. In this study we address the choice of a suitable structural template for the homology modeling of the P2Y12R. A sequence analysis of the human (h) P2Y12R sequence and the GPCR available in the Protein Data Bank (PDB) database (http://www.rcsb.org/pdb/) suggested the crystal structure of the CXCR4 as the most similar structure among the currently available GPCR structures. In order to clarify if a slightly higher sequence identity between the P2Y12R and CXCR4, compared to the other templates, could provide an improved homology model of the P2Y12R, we compared the binding cavities of three different P2Y12R homology models, generated using the RHO (PDB ID: 1F88), the A2AAR (PDB ID: 3EML), and CXCR4 (PDB ID: 3ODU) structures as templates. We compared the structural features of the models and we identified specific residues of interest in the binding cavities that have never been considered in previous modeling.
Results and discussion
Sequence analysis and homology modeling
A sequence analysis of the P2Y12R and the current solved GPCRs was conducted to understand the degree of similarity between our target and the possible templates. The P2Y12R sequence was aligned with the sequences of the GPCR structures retrieved from the PDB, being cautious to correctly align the transmembrane (TM) regions using the residues that are highly conserved among the Class A family of GPCRs and not allowing insertions or deletions in these domains. The alignment of the loop regions connecting the transmembrane helical domains (TMs) is a challenging aspect due to the different lengths of the loops and the low sequence identities between the different GPCRs. Fig. S1 (Supporting Information) shows a multiple alignment of the P2Y12R and the GPCRs having an x-ray structure. Table 1 summarizes the percent identity of the aligned sequences of P2Y12R and the considered GPCR structures. Considering the complete receptor sequence, the identities are low with values between 15.5 and 21.9%, and with only P2Y12R and CXCR4 having >20% identity. However, if only the TMs are considered during the alignment the identity percentages between the target sequence and the other GPCRs are higher, with values from 18.8%, between P2Y12R and RHO, to 25.6%, between P2Y12R and CXCR4.
However, a slightly higher sequence identity between P2Y12R and CXCR4 alone cannot justify the choice of the CXCR4 structure as the optimal template for the homology modeling of our target. Therefore, we examined whether the sequence similarities between P2Y12R and CXCR4 could be reflected in common structural features and whether the unique structural features of the novel CXCR4 structure could be applied to modeling of the P2Y12R to achieve an improved 3D structure with new structural insights to be used in drug design.
The homology model of the hP2Y12R was generated using the recently disclosed crystal structure of the CXCR4 as a template [11]. The model was optimized focusing mainly on the binding cavity of the receptor, the putative site of nucleotide binding. A flexible molecular docking was conducted with the non-selective P2Y12R agonist 2MeSADP 2 or the selective P2Y12R antagonist PSB-0739 3 (Fig. 1) in order to find their possible binding modes in the optimized binding pocket of the model. The putative binding cavity of the CXCR4-based model (denoted P2Y12R-CXC) was compared with the binding pockets of previously built P2Y12R models obtained by means of homology modeling based on RHO [17] and the A2AAR crystal structure [3]. A superposition of three models of the P2Y12R based on different templates is shown in Fig. 2. The structural features that differ in the three models were outlined and discussed. The residue side chains forming the binding cavity in each model were analyzed to clarify their role in the characterization of the pocket and in the binding of small ligands.

Superimposition of three models of the P2Y12R based on different GPCR structural templates. The TM domains of P2Y12R-RHO (red), P2Y12R-A2A (blue), and P2Y12R-CXC (green) were superimposed by the Cα atoms of the residues. Panel a side view of the Cα ribbon of the P2Y12R models with the extracellular side on the top of the panel. Panel b the upper part of TMs 2,3,4, and 5, and EL1 and EL2 are shown viewed from the extracellular side highlighting the different conformation assumed by EL1 and EL2 in the three P2Y12R models. Panel c the extracellular regions of TM6, TM7 and EL3 are shown from the side view. In P2Y12R-CXC, TM7 is two α-helical turn longer and EL3 is shorter compared to TM7 and EL3 in P2Y12R-RHO and P2Y12R-A2A
P2.58 and the TM2 secondary structure
The multiple sequence alignment of the P2Y12R against the other GPCRs revealed similarities of our target with the CXCR4 that were not seen with the other templates. The P2Y12R and CXCR4 share a common proline residue in the second TM (TM2) domain. Proline, which lacks the amide proton normally present in other amino acids that serves to stabilize the alpha helical conformation by H bonding, can break the periodicity of the alpha helix causing kinks and bulges in the helical domains. The chemokine receptors and P2Y receptors share a proline at position 2.58 (using the nomenclature of Ballesteros and Weinstein [18]), while the other available templates have Pro2.59, e.g., ADRB1, ADRB2, A2AAR, DRD3, or no proline at all, e.g., RHO. On the other hand, a particularity in the sequences of the P2Y12R subfamily of purinergic receptors (including also P2Y13 and P2Y14 receptors) is the lack of the highly conserved proline of TM5 (5.50). However, this conserved proline is present in the other receptors of the P2Y1 subfamily. In this case, an unambiguous alignment of TM5 of the P2Y12R was achieved using the second most common residue of the helix, Tyr5.58.
The proline residue in TM2 of the P2Y12R is not conserved among the Class A family of GPCRs. The structural consequences of a proline on the conformation of TM2 have been studied in depth for several other GPCRs, especially regarding the homology modeling of those receptors [19–21]. The most conserved residue in TM2 of Class A GPCRs is an aspartic acid, Asp2.50, which normally is used to guide the sequence alignment of TM2. In humans, about 40% of the Class A GPCRs have a proline at position 2.58, such as the chemokine receptors or the P2Y receptors, while 37% have a proline at 2.59, such as the amine receptors and the ARs. Only 21% of the Class A GPCRs, such as rhodopsin, lack a proline in TM2, and less than 5% have a Pro2.60 [21]. Prior to the determination of the CXCR4 structure, the available GPCRs structures revealed the presence of a bulge and a kink in TM2. In the case of the amine receptors and the A2AAR, this bulge deviated from the canonical helical periodicity with a helical turn containing five amino acids instead of the normal four. In these receptors, Pro2.59 is located at the C-terminus of the bulge elbow and at the center of the helical kink of TM2. RHO, on the other hand, has no proline in TM2. However, the RHO structure showed a similar bulge and kink in this helix (Fig. 3a). The helical bulge is caused by the helix-distorting GGXTT motif, due to the presence of two adjacent glycine residues, Gly89(2.56) and Gly90(2.57). As a consequence of this helical bulge in TM2 the residue at position 2.59, Pro61 in the A2AAR and Thr92 in RHO, was located on the lipid face of TM2, while the preceeding residue, Ile60 in A2AAR and Phe91 in RHO, was located at the TM1–TM2 interface. The same orientation was maintained by the corresponding residues in the RHO- or A2AAR-based models of the P2Y12R (denoted P2Y12R-RHO and P2Y12R-A2A). Thus, residue Phe79 at position 2.59 was exposed to the lipid environment, and Pro78(2.58) was located at the TM1–TM2 interface (Fig. 3b, c). Moreover, in P2Y12R-RHO and P2Y12R-A2A the bulky residue Phe77(2.57), corresponding to the small residues Gly90 of RHO and Ala59 of A2AAR, was at the interface between TM2 and TM3 where the two helices cross, creating a steric clash with the comparably bulky side chain of Ile103(3.31).

Comparison of the structure of TM2 in the P2Y12R models and the templates. Panel a the Cα traces of TM2 in the crystal structures of RHO (cyan), A2AAR (green), and CXCR4 (magenta) are shown with the N-terminus of the helix at the top of the panel. TM2 is characterized by a kinked structure due to the presence of the GGXTT motif in RHO (the two glycine residues are labeled), Pro2.59 in A2AAR, and Pro2.58 in CXCR4. In RHO and A2AAR, but not in CXCR4, TM2 shows a bulged elbow with 5 residues in one helical turn instead of 4 residues. The backbone atoms of the residues preceding and following the kink are shown. Panel b, c, d: the Cα ribbon of TM2 in the context of TM1 and TM3 is shown for P2Y12R-RHO (b), P2Y12R-A2A (c), and P2Y12R-CXC (d). The TM domains are shown with the extracellular side at the top of the panel and labeled as indicated. Pro5.58 is indicated with the surrounding residues of TM2. In P2Y12R-RHO (b) and P2Y12R-A2A (c), Pro78 is located in the interface between TM1 and TM2, while in P2Y12R-CXC (d) the Pro78 is facing the lipid environment. Where TM2 and TM3 cross, the residues are represented also as dotted surface. In P2Y12R-RHO and P2Y12R-A2A, there was a clear steric clash between the bulky Ile103 in TM3 and Phe77 in TM2. In P2Y12R-CXC, instead, the small Thr76 fit properly in the junction between TM2 and TM3
A completely different helical conformation of the C-terminus of TM2 was revealed in the recently determined structure of CXCR4. Govaerts et al. [22, 23] proposed that this sequence motif was key to shape the binding pocket of chemokine receptors. Deville et al. [21] predicted that the T(S)XP motif in TM2 of CXCR4 is incompatible with a helical bulge found in the GPCR structures reported prior to CXCR4. Pro2.58 of CXCR4 creates a kink in TM2 that bends the extracellular part of the helix toward the center of the receptor, but does not cause a distorted turn involving five residues (Fig. 3a). The wobble angle of the C-terminal region of TM2 in the CXCR4 structure is different from the angles in the previously reported structures. In the CXCR4 structure the side chain of residue 2.59, Phe93, is still exposed to the lipid environment, as is Pro92(2.58). At the TM1-TM2 interface, where residue 2.58 is found in the RHO and A2AAR, the side chain of Leu91(2.57) is now located. Pro78(2.58) of the model was facing the lipids as well as Phe79(2.59), while the bulky Phe77(2.57) was located at the TM1–TM2 interface and no longer facing Ile103(3.31) on TM3. In this new P2Y12R model, in the region where TM2 and TM3 cross, the bulky Ile103(3.31) was positioned opposite the small side chain of Thr76(2.56), and thus avoiding the steric clash with Phe77 observed in the previous models (Fig. 3).
Extended helix 7 and disulfide bridge between EL3 and N-terminus
The role of the extracellular loops (ELs) in ligand recognition within the P2Y family was first demonstrated by site-directed mutagenesis of the human P2Y1 receptor [14]. TM7 in the CXCR4 structure is extended by two helical turns in the extracellular direction compared to the structures of RHO or A2AAR, and the cysteine residue involved in the disulfide bridge with the N-terminus is at the C-terminal end of this helix. It is unclear whether the longer helical conformation of TM7 in CXCR4 is a consequence of the structural constraint by the disulfide bridge with the N-terminus, since this is the first crystal structure having this feature. There is no direct evidence suggesting that the P2Y12R should assume a similarly extended TM7 as in the CXCR4 template. However, the sequence alignment of these two receptors showed a very high similarity in this region. EL3 is of variable length within the GPCR superfamily, as in the GPCR structures considered in this paper. Counting the residues between positions 6.60 and 7.32, the last and the first amino acids of TMs 6 and 7 in RHO, EL3 consists of a short sequence of 7 or 8 residues in RHO and A2AAR, respectively, or of a longer sequence of 16 residues in CXCR4. The EL3 in the P2Y12R has a sequence length of 15 residues, comparable to the CXCR4. Comparing the nature of these 15 residues, we found a high similarity between the sequence of P2Y12R and CXCR4 (Table 1, Fig. S1 of Supporting Information), especially in the residues following Cys7.25. This suggests that the EL3 secondary structure in the P2Y12R might also assume a conformation similar to CXCR4. Moreover, for the P2Y12 receptor a disulfide bridge between Cys17 in the N-terminal and Cys270 in EL3 may also exist, although it does not seem to be essential for receptor function [24].
In P2Y12R-CXC, in contrast to the RHO- and A2AAR-based models, the alpha helical structure of TMs 6 and 7 were of equal length to the template structure, making the EL3 much shorter than in the other models (Fig. 2c). These particular features of the TM6, TM7 and EL3 created a more extended and more solvent-exposed binding cavity in P2Y12R-CXC, especially if compared to P2Y12R-RHO. In that model, the region of the binding cavity formed by residues from the upper part of TM6 and TM7 in P2Y12R was not accessible to the binding pocket due to shielding by the deep EL2.
Binding cavities and ligand poses in the P2Y12R models
The native nucleotide agonist ADP 1 was not docked in our study, in preference to the closely related 2-methylthio analogue 2, which is roughly 3 orders of magnitude more potent at the a P2Y12R [25, 26]. We recognize that the template for this homology modeling is an inactive receptor. However, a recent example demonstrates that docking of agonists to basal-state homology models can be highly predictive of a crystallographic structure of an agonist-bound GPCR [13, 27]. It could be instructive to dock the agonist in a P2Y12R model based on the new active state structures, although this would not have the advantages of the similarity to the CXCR4 structure.
The reversible antagonists that contained an adenine or 8-azaadenine moiety, i.e., nucleotide derivative 4 and the nucleoside analogues 5 and 6, were not docked in the receptor. However, we studied the possible docking modes of the potent heterocyclic, reversible antagonist 3 [28, 29], the leading compound of the piperazinyl-glutamate-pyridine series 7 [30], and the irreversible antagonist 8 [25].
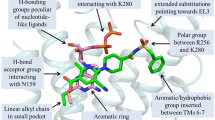
Key residues for the binding of ligands in the binding cavity of the P2Y12R were previously identified by site-directed mutagenesis. Hoffmann et al. demonstrated that the charged residue Arg256(6.55) coordinated the negative charged phosphate groups of the non-selective agonist 2MeSADP 2 [31] and the sulfonic acid residue at ring D (refer to Fig. 1) of the selective antagonist PSB-0739 3 [28]. Lys280(7.35) and Tyr259(6.58) were also involved in the recognition of the ligands. The mutation of these two residues led to a decrease of the binding of ADP and 2MeSADP [31]. The docked poses of 2MeSADP 2 and PSB-0739 3 in the binding pocket of P2Y12R-CXC agreed with previously published site-directed mutagenesis studies of indicating a role of Arg256 and Lys280 (Fig. 4a, b). Both the guanidinium group of Arg256 and the amino group of Lys280 anchored the β-phosphate group of 2MeSADP. One helical turn above Arg256 in TM6, the hydroxyl group of Tyr259 interacted with the α-phosphate group of the agonist. In the docked binding mode of PSB-0739 Arg256 coordinated the sulfonic acid residue in ring D of the antagonist, while Lys280 interacted with the aromatic ring D itself through a cation–π interaction. The aromatic ring of Tyr259 was involved in a π–π stacking with ring E. Other residues that formed the binding cavity in P2Y12R-CXC were hydrophobic and aromatic residues from TM1, TM3, TM7, and EL2 including Leu284(7.39), Phe104(3.32) and Tyr105(3.33) at the bottom of the pocket, Tyr32(1.39), and Phe177 in EL2. The hydroxyl group of Tyr105 stabilized the 3′-OH on the ribose ring of 2MeSADP and the sulfonic acid in ring D of PSB-0739. The amino group at position 6 of 2MeSADP interacted with the side chain of Asp84(2.63). Residues from EL2 were also involved in the binding of the ligands. The aromatic ring of Phe177 anchored the aromatic cores of both 2MeSADP and PSB-0739. Moreover, in the docking pose of PSB-0739 the sulfonic group in ring C was coordinated by the backbone amino group of Phe177 and the side chain group of Lys174. The docking pose of PSB-0739 in the binding cavity of P2Y12R-CXC was in agreement with the structure–activity relationships of the anthraquinone derivatives recently investigated by Baqi et al. [29]. The π–π stacking between ring E of PSB-0739 and Tyr259(6.58) can explain the critical role of this aromatic ring for the potency of the anthraquinone series. Moreover, the docking mode of PSB-0739 in P2Y12R-CXC raise the hypothesis of a negatively charged sulfonate or carboxylate group at position 2 of the anthraquinone moiety to create a strong interaction with the Lys174 side chain. The same key residues involved in the interactions with PSB-0739 and 2MeSADP in the docked poses to P2Y12R-CXC were found crucial for the stabilization of the docked compound 7 (Fig. S3) [30]. The aromatic ring of the pyridine moiety was anchored by a π–π interaction with Phe177 (EL2) on one side of the pocket and by a cation–π interaction with Lys280 on the other side. Moreover, the γ-carboxylate of the glutamate chain interacted with the positive charges of both Arg256 and Lys280.

Binding modes of agonist 2MeSADP 2 (a) and reversible antagonist PSB-0739 3 (b) (structures shown in Fig. 1) in P2Y12R-CXC. The ligands occupy a pocket in the upper part of the TM bundle and are embedded by residues from TM1, TM2, TM3, TM6, TM7 and EL2. The key residues in the binding pocket are shown, and the H bond interactions are depicted as dashed lines. The residues in the putative binding pocket are shown in stick with C-atoms colored in gray; the ligands are in ball-and-stick representation with the carbon atoms colored in magenta for 2MeSADP 2 and green for PSB-0739 3. The helices are sequentially color coded from blue (TM1) to red (TM7)
The docking poses of 2MeSADP in P2Y12R-RHO and P2Y12R-A2A revealed a similar role of the key residues Arg256(6.55), Tyr259(6.58) and Lys280(7.35). Nevertheless, the different conformations of the extracellular loops in the three models led to binding cavities with different characteristics (Fig. 5a–c). In P2Y12R-RHO, the deep conformation of EL2 restricted the agonist within a deep cavity between TM1, TM2, TM3 and TM7. The aromatic core of 2MeSADP was anchored by π–π stacking with the phenyl ring of Phe104(3.32), very low in the pocket compared with the 2MeSADP docking pose in P2Y12R-CXC, where Phe104(3.32) defined the bottom of the pocket below the nucleotide. The more open conformation of EL2 in P2Y12R-A2A allowed the ligand to adjust to a position in a cavity that was higher in the TM bundle compared to the binding pocket in P2Y12R-RHO (Fig. 5). Nevertheless, the shape of the binding cavity in P2Y12R-A2A was severely influenced by the particular conformation of EL1 and EL2. In the A2AAR crystal structure there were three disulfide bridges that constrain EL2 with the extracellular region of TM3 and EL1. One disulfide bond was the canonical bridge between Cys77(3.25) in TM3 and Cys166 in EL2, common to almost all the family A GPCRs. The two additional disulfide bridges occurred between Cys71(EL1) and Cys159(EL2), and between Cys74(EL1) and Cys146(EL2), and are unique to the A2AAR. In the crystal structure, these two disulfide bridges, besides conferring structural rigidity to the loops, caused a shift of EL1 toward EL2 and consequently shifted the C-terminus of TM2 toward the center of the receptor. Also, the N-terminus of TM3 was shifted toward TM4, relative to other GPCR structures (Fig. 3). The co-crystallized antagonist in the A2AAR structure binds in a cavity that is far from TM2, and the particular orientation of this domain does not influence the binding of the ligand. In contrast, the P2Y12R binding pocket is very close to TM2, as suggested by the docked poses of 2MeSADP in the three P2Y12R models. In P2Y12R-A2A, the shift of TM2 toward the binding cavity pushed the ligand into a small pocket between TM1 and TM7. The docked pose of 2MeSADP in this model showed interactions between the N1 nitrogen of the adenine ring and the side chain of Tyr32(1.39) inTM1, and the 2-methylthio group of the agonist was in proximity to residues such as Pro78(2.58), Leu284(7.39), Thr287(7.42), and Ser288(7.43). Instead, in P2Y12R-CXC the different orientation and characteristic kink of TM2 left more space in the cavity between TM2 and TM7, allowing the docked nucleotide to interact with residues from TM2, such as the carboxyl group of Asp84(2.64). Tyr32(1.39) still defined the binding pocket in P2Y12R-CXC, but it was further from the docked agonist than in other P2Y12R models without directly interacting with the molecule (Fig. 4).

Comparison of the binding cavities in the three models of the P2Y12R. The molecular surfaces of the binding pocket in the models are shown with the docked 2MeSADP 2. Panel a binding pocket of P2Y12R-RHO; panel b binding cavity of P2Y12R-A2A; panel c binding cavity of P2Y12R-CXC. The helices are sequentially color coded from blue (TM1) to red (TM7)
Exposure of the conserved disulfide bridge to the binding cavity
P2Y12R-CXC showed another particular structural feature that was not manifest in the other two models. The particularly open β-sheet conformation of EL2 in the CXCR4-based model allowed the exposure of the cysteine residues involved in the highly conserved disulfide bridge between the upper part of TM3 and EL2. This feature is especially interesting regarding the P2Y12R. The thienotetrahydropyridine drugs, i.e., Clopidogrel and Prasugrel, are selective irreversible blockers of this receptor. After bioactivation by the cytochrome P450 system, the free thiol metabolites (e.g., 8 for Clopidogrel) covalently bind to the receptor [32]. Mutagenesis experiment with the active metabolite of Prasugrel by Algaier et al. suggested the interaction of the thienopyridine active metabolites with Cys97(3.25) and Cys175 (EL2) of the conserved disulfide bridge of P2Y12R [33]. In P2Y12R-RHO, the cysteine residues of the conserved disulfide bridge were shielded by residues of the EL2 (Fig. S2). In P2Y12R-A2A, the corresponding cysteines are even more covered, not only by residues of EL2 but also by residues from TM2 and more shifted toward the center of the receptor. In P2Y12R-CXC, instead, Cys175 in EL2 was more accessible from the binding cavity, partially covered by the side chain of Phe177, and in close proximity to the docked ligands.
To test the accessibility of the conserved disulfide bridge from the binding pocket of P2Y12R-CXC, we docked the irreversibly-binding, active metabolite diastereoisomers 8 of the clinically utilized antagonist Clopidogrel. Both the 4S and 4R diastereoisomers bound in the so-called minor binding pocket [34], a region of the binding crevice located between TMs 1, 2, 3, and 7. In the binding of the Clopidogrel metabolites, there is no involvement of key residues such as Arg256(6.55) and Lys280(7.35). The major anchoring of both the diastereoisomers of 8 to the receptor consisted of a salt bridge between the carboxyl group of the derivatives and the positively charged side chain of Lys174 in EL2. The same carboxyl group is also stabilized by an H bond interaction with the backbone amino group of Phe177 (EL2). In the docked pose of the 4S diastereoisomer 8b, the free thiol group is found in close proximity (with a distance of about 4.7 Å) to the sulfur atom of Cys175, as shown in Fig. 6. In spite of the same strong interaction between the carboxyl group of the metabolite and the Lys174 side chain, the thiol group of the 4R diastereoisomer of 8 was unable to reach the disulfide bridge. The accessibility of Cys175 from the binding pocket in the docked pose of 8b was possible because of the rotation of the phenyl ring of Phe177 that in the P2Y12R-CXC complexes with 2MeSADP and PSB-0739 was interacting with the aromatic cores of the ligands. This interaction occurred through π-π stacking, which shielded the disulfide bridge from these reversibly-binding ligands. From the docking results we speculate that the active metabolite of Clopidogrel has to assume an S conformation at position 4 to break the conserved disulfide bridge and irreversibly form a new disulfide linkage with Cys175 in EL2.

Docking poses of the active metabolite diastereoisomers 8 of Clopidogrel in P2Y12R-CXC. Panel a the proposed binding mode of 8b in the binding pocket of P2Y12R-CXC. The ligand is anchored in the binding pocket by the interactions of the carboxyl group of the pyridine derivative and the side chain of Lys174 and the backbone amino group of Phe177 in EL2. The free thiol group of 8b is in close proximity with the sulfur atom of Cys175 of the conserved disulfide bridge between EL2 and TM3. Panel b proposed binding pose of the 4R diastereoisomer of Clopidogrel. The carboxyl group of the pyridine derivative is involved in a similar interaction with Lys174, but the free thiol cannot reach the disulfide bridge. The receptor residues are depicted in stick colored by element with the carbon atoms in gray. The docked compounds are represented as ball-and-stick colored by element with the carbons in magenta. The key interactions between the ligands and the residues in the pocket are shown as dotted lines
Conclusions
In conclusion, we have selected a CXCR4-based model of the P2Y12R as being the most consistent with observed pharmacology and mutagenesis studies and with the docking of the both reversibly and irreversibly-binding small molecules. The three models of the P2Y12R based on different template structures were all able to show the key roles of important residues such as Arg256(6.55), Tyr259(6.58), and Lys280(7.32) in ligand coordination. However, each model showed limitations in describing the structural features of the binding cavity in the P2Y12R. The most evident structural differences of the binding pocket in the three models were located mainly in TM2 and the extracellular domains. In P2Y12R-RHO, the pocket was deeply buried within the TM bundle, while in P2Y12R-A2A the ligand occupied a small pocket between TM1 and TM7 due to the particular orientation of TM2 more proximal to the center of the receptor. P2Y12R-CXC, with the novel orientation of the kinked TM2, was able to highlight new residues of TM2 that were not previously predicted to be in the binding cavity, such as Lys80 and Asp84. Moreover, the CXCR4 structure is the only currently available GPCR structure that shares with the P2Y12R a disulfide bridge between the N-terminal domain and EL3, making this a more suitable template for this domain of the P2Y12R. P2Y12R-CXC also illustrated the accessibility of the conserve disulfide bridge in TM3 and EL2 to the binding cavity, suggesting that the thienopyridine antithrombotic drugs, e.g., the active metabolite(s) of Clopidogrel, can bind in the same binding cavity here described. The free thiol group of the 4S diastereoisomer, but not the 4R isomer, was found in close proximity (~4.7 Å) to the sulfur atom of a disulfide bridge involving Cys175. Therefore, we predict that the 4S isomer is the more active diastereomer for covalent binding. The new insight into the structural features of the P2Y12R from the new model based on the recently published CXCR4 structure should be validated by site-directed mutagenesis and other experimental studies. Nevertheless, we conclude that the CXCR4 structure is a more suitable template for the modeling of the P2Y12R compared to other GPCR templates.
Supporting information available
Methodology for sequences analysis and multiple sequence alignment, homology modeling and docking; the multiple alignment of P2Y12R and other GPCRs; a figure showing the accessibility of the disulfide bridge between Cys97(3.25) and Cys175 from the binding cavity in the three P2Y12R models; a figure showing the docking pose of compound 7 in the binding cavity of P2Y12R-CXC.
References
Dorsam RT, Kunapuli SP (2004) Central role of the P2Y12 receptor in platelet activation. J Clin Invest 113:340–345
Giossi A, Pezzini A, Del Zotto E, Volonghi I, Costa P, Ferrari D, Padovani A (2010) Advances in antiplatelet therapy for stroke prevention: the new P2Y12 antagonists. Curr Drug Targets 11:380–391
Jacobson KA, Deflorian F, Mishra S, Costanzi S (2011) Pharmacochemistry of the platelet purinergic receptors. Purinergic Signal. doi:10.1007/s11302-011-9216-0 (in press)
Moro S, Hoffmann C, Jacobson KA (1999) Role of the extracellular loops of G protein-coupled receptors in ligand recognition: a molecular modeling study of the human P2Y1 receptor. Biochemistry 38:3498–3507
Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289:739–745
Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454:486–491
Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265
Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK (2007) Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450:383–387
Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, IJzerman AP, Stevens RC (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217
Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071
Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095
Scheerer P et al (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455(7212):497–502
Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka KB (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469:175–180
Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC (2011) Structure of an agonist-bound human A2A adenosine receptor. Science. doi:10.1126/science.1202793 (in press)
Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA (2005) Human P2Y6 receptor: molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J Med Chem 48:8108–8111
Bissantz C, Bernard P, Hibert M, Rognan D (2003) Protein-based virtual screening of chemical databases. II. Are homology models of G-protein coupled receptors suitable targets? Proteins 50:5–25
Costanzi S, Mamedova L, Gao ZG, Jacobson KA (2004) Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem 47:5393–5404
Ballesteros J, Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 25:366–428
Langelaan DN, Wieczorek M, Blouin C, Rainey JK (2010) Improved helix and kink characterization in membrane proteins allows evaluation of kink sequence predictors. J Chem Inf Model 50:2213–2220
Zhang R, Hurst DP, Barnett-Norris J, Reggio PH, Song ZH (2005) Cysteine 2.59(89) in the second transmembrane domain of human CB2 receptor is accessible within the ligand binding crevice: evidence for possible CB2 deviation from a rhodopsin template. Mol Pharmacol 68:69–83
Devillé J, Rey J, Chabbert M (2009) An indel in transmembrane helix 2 helps to trace the molecular evolution of class A G-protein-coupled receptors. J Mol Evol 68:475–489
Govaerts C, Blanpain C, Deupi X, Ballet S, Ballesteros JA, Wodak SJ, Vassart G, Pardo L, Parmentier M (2001) The TXP motif in the second transmembrane helix of CCR5. A structural determinant of chemokine-induced activation. J Biol Chem 276:13217–13225
Govaerts C, Bondue A, Springael JY, Olivella M, Deupi X, Le Poul E, Wodak SJ, Parmentier M, Pardo L, Blanpain C (2003) Activation of CCR5 by chemokines involves an aromatic cluster between transmembrane helices 2 and 3. J Biol Chem 278:1892–18903
Savi P, Zachayus J-L, Delesque-Touchard N, Labouret C, Hervè C, Uzabiaga M-F, Pereillo J-M, Culouscou J-M, Bono F, Ferrara P, Herbert J-M (2006) The active metabolite of Clopidogrel disrupts P2Y12 receptor oligomers and partitions them out of lipid rafts. Proc Natl Acad Sci USA 103:11069–11074
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB (2001) Identification of the platelet ADP receptor targeted by anti-thrombotic drugs. Nature 409:202–207
Ivanov AA, Barak D, Jacobson KA (2009) Evaluation of homology modeling of G protein-coupled receptors in light of the A2A adenosine receptor crystallographic structure. J Med Chem 52:3284–3292
Hoffmann K, Baqi Y, Morena MS, Glänzel M, Müller CE, von Kügelgen I (2009) Interaction of new, very potent non-nucleotide antagonists with Arg256 of the human platelet P2Y12 receptor. J Pharmacol Exp Ther 331:648–655
Baqi Y, Atzler K, Köse M, Glänzel M, Müller CE (2009) High-affinity, non-nucleotide-derived competitive antagonists of platelet P2Y12 receptors. J Med Chem 52:3784–3793
Parlow JJ, Burney MW, Case BL, Girard TJ, Hall KA, Harris PK, Hiebsch RR, Huff RM, Lachance RM, Mischke DA, Rapp SR, Woerndle RS, Ennis MD (2010) Piperazinyl glutamate pyridines as potent orally bioavailable P2Y12 antagonists for inhibition of platelet aggregation. J Med Chem 53:2010–2037
Hoffmann K, Sixel U, Di Pasquale F, von Kügelgen I (2008) Involvement of basic amino acid residues in transmembrane regions 6 and 7 in agonist and antagonist recognition of the human platelet P2Y12-receptor. Biochem Pharmacol 76:1201–1213
Collet JP, Montalescot G (2009) P2Y12 inhibitors: thienopyridines and direct oral inhibitors. Hämostaseologie 29:339–348
Algaier I, Jakubowski JA, Asai F, von Kügelgen I (2008) Interaction of the active metabolite of prasugrel, R-138727, with cysteine 97 and cysteine 175 of the human P2Y12 receptor. J Thromb Haemost 6:1908–1914
Rosenkilde MM, Benned-Jensen T, Frimurer TM, Schwartz TW (2010) The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol Sci 31:567–574
Acknowledgments
We thank Dr. Stefano Costanzi (NIDDK) and Dr. David Erlinge (Lund University, Lund, Sweden) for helpful discussion. This research was supported by the Intramural Research Program of the NIH, NIDDK.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Deflorian, F., Jacobson, K.A. Comparison of three GPCR structural templates for modeling of the P2Y12 nucleotide receptor. J Comput Aided Mol Des 25, 329–338 (2011). https://doi.org/10.1007/s10822-011-9423-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-011-9423-3