
Abstract
AllChem is a system that is intended to make practical the generation and searching of an unprecedentedly vast number (∼1020) of synthetically accessible and medicinally relevant structures. Also, by providing possible synthetic routes to a structure along with its design rationale, AllChem encourages simultaneous consideration of both costs and benefits during each lead discovery and optimization decision, thereby promising to be effective with synthetic chemists among its primary users. AllChem is still under intensive development so the following initial description necessarily has more the character of an interim progress report than of a finished research publication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Based on all currently available information, what should be the next compound(s) to make and test? Addressing that critical and recurring question is the goal of almost all CAMD methodology. As with all investments and wagers, any make-and-test decision has both a possibly beneficial outcome and a relatively certain cost. But CAMD development has focused almost exclusively on outcomes, leaving most issues of cost to the laboratory chemist.
The AllChem™ project seeks to build CAMD methodology that simultaneously addresses costs as well as benefits, and on an unprecedentedly large scale. The lecture on which this article is based [1] represented the first formal disclosure of the AllChem technology. Its major components are:
-
A collection of roughly 5 × 106 synthons (herein “synthon” denotes a structure with one or more open valences each having a defined reactivity), which must combinatorially represent as many as (5 × 106)3 or 1020 complete structures having a general topology of A-B-C.
-
The program gensyn, which generates synthons by recursively applying reactions to building blocks, under various pragmatic constraints. The 5 × 106 synthons result from ∼100 reactions and ∼7,000 building blocks. To the best of our knowledge [2–22], such a process has not previously been implemented, at least on any comparable scale.
-
Suitably effective and rapid filtering/searching techniques, in particular the topomer methodologies for identifying novel and attractively shape similar or shape superior structures, described elsewhere [23].
These components are united by means of a relational database (Oracle), including a user interface that is intended for laboratory as well as computational chemists. The topomer searching methodologies seem robust and automatic enough for laboratory chemists to use effectively in choosing the most biologically promising structures (recognizing that the complementary judgments about synthetic costs will almost always be made by laboratory chemists).
Initially the target laboratory chemist-users have been those working at Tripos Discovery Research (TDR) in Bude, Cornwall, UK. As suppliers of outsourced chemistry services, these chemists have had specific application priorities that have been emphasized in AllChem development:
-
LeadHopping™, the identification of otherwise dissimilar ligand structures that are shape similar and therefore probably biologically similar to a “lead” or query structure. A “topomer”-based shape similarity technology has repeatedly proven effective for lead hopping [24].
-
Structurally novel scaffolds with properties appropriate for general screening libraries to be synthesized combinatorially, for example small size and high “interest.”
Performance goals for AllChem’s development include:
-
Complete search times should not be longer than overnight.
-
The vast majority of the synthetic routes that generate the synthons should seem plausible to laboratory chemists.
-
Considering the dynamic quality of synthetic knowledge, the time to recreate or otherwise edit the synthon collection should be no greater than a week.
AllChem is intended as a successor to ChemSpace [25, 26], which has for 10 years provided TDR with similar access to ∼1014 structures by manipulating conventional combinatorial libraries, composed of chemist-proposed scaffolds and commercially offered side chains linked together by standard combinatorial chemistry protocols. The most important ChemSpace limitation is that most structures found in the medicinal chemistry literature are not such simple assemblies. Although the majority of such literature syntheses are still formally combinatorial (composed of two or three large pieces connectable in principle by combinatorial processes), usually at least one of the pieces is not available commercially, instead being built up in several steps. Such structures have far more novelty, including a higher density of the polar features likely to produce higher ligand efficiency. Only about 5% of structures found in the medicinal chemistry literature are included within ChemSpace virtual libraries, whereas it is hoped that around 50% of published structures may be identifiable by AllChem searches.
Methods
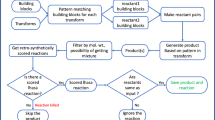
Figure 1 attempts to convey a high-level understanding of AllChem processes and architecture. In its center, bisected by a dotted line, is the collection of Synthons. Everything shown above that dotted line involves Generating those synthons and everything below supports their Searching.

Summary of the AllChem processes for generating and searching very large databases. See text for details
Synthons are produced by the gensyn program, which in a typical run applies a set of 100 reactions successively to each of 7,000 building blocks (B). Each resulting structure becomes another building block (B1, B2, -B, -B-), constituting a recursive process with its individual sequences being bounded mostly by the maximal “cost” for any synthetic route. Those structures which have open valences are added to the synthon collection. All new structures are added to the Intermediates table, to support the display of synthetic routes. Included but not shown in Fig. 1 is a Reactions table, which records every gensyn conversion of one structure into another. Also not shown are reactions that combine selected synthons with other selected synthons, “bimolecular” reactions that significantly increase overall structural diversity.
It is expected that searching among the 1020 complete structures representable by combining these synthons will (and probably must) always begin by discarding almost all the candidates, using the topomer principles of shape similarity. Usually this filtration would be explicit, for example when leadhopping from a query structure as shown in Fig. 1. The query structure (here Z-Y-X-W) is then fragmented in all reasonable ways, by breaking its acyclic single bonds individually and pairwise. Each of the fragments comprising the resulting pairs and triplets is converted into a topomer, to be shape-compared with every one of the stored synthons. (Thus by replicating the synthon data base these comparisons could be trivially, though unnecessarily, parallelized.) The vast majority of stored synthons are by themselves so shape-dissimilar from a query fragment as to eliminate every complete structure that includes that synthon. (This “branch-and-bounds” behavior is the fundamental reason that topomer similarity can completely search such large structural spaces. The topomer similarity comparisons are also themselves very fast, tens of thousands per second.) To be acceptable, a product structure must also have been formed by joining open valences that have complementary reactivity.
Addressing the other TDR application need that for novel scaffolds, requires only conventional searches of the large synthon collection itself and so is not shown in Fig. 1. Other synthon data bases help address this need, for example by allotting more resources to reaction sequences that seem more likely to form additional rings. (Of course, the size and composition of a particular synthon data base, though entirely deterministic in principle, are in practice highly dependent on the building blocks, reactions, and other run-time parameters provided to gensyn.)
Synthon generation
The most challenging new aspect of the AllChem system has been the design and implementation of gensyn. A major concern is not to waste the most expensive system resource, the time and attention of highly trained synthetic chemists, by generating too many reaction sequences that are either implausible before or, far worse, unworkable after, further literature or experimental exploration. Yet our cumulative synthetic chemistry knowledge base is almost anecdotal in its sparseness, compared to the immensity of structures and their potential reactions, while the effort needed to fully assemble and curate even a few reaction instances from that knowledge base is completely beyond the scope of this project. And of course it is also impossible to evaluate more than a few of the over 107 reaction steps produced by a typical gensyn run as shown in Fig. 1.
Despite this challenging combination of constraints, designer and user satisfaction with gensyn’s output is currently high. So it seems appropriate to list some of its more important embedded design and implementation decisions.
-
All information describing every individual reaction, in particular its scope and limitations, is taken from external text files that are directly manipulable by humans [27].
-
The included reactions are those already successfully practiced by TDR chemists, augmented by a few heterocycle-forming sequences taken from the current medicinal literature (in the quest for novel scaffolds [28]). Usually the building blocks are simply those having the highest “price/supplier scores,” this score being a rough composite of cost including vendor responsiveness and any regulatory issues.
-
Scope constraints on any specific reaction result mostly from inspection of random samples of 50–100 individual applications of that reaction taken from a production run of gensyn. It is desired that at least 90% of these proposed specific reaction instances should seem unchallengeable. Conversely, undue restrictions on scope are prevented by collecting interesting reaction sequences from the literature, with gensyn being required in regression testing to regenerate each final structure given the starting structure(s).
-
Reactive intermediates are explicitly represented. For example, the generic amide formation reaction requires three steps:
$$ {\text{RC}}\left( {{\text{ = O}}} \right){\text{Cl}} \to {\text{RC}}\left( {{\text{ = O}}} \right){\text{ - }}\left[ {\text{e}} \right] $$$${\rm R}^{\prime}{{\rm NH2}}\to {\rm R}^{\prime}{\rm NH - }\left[ {\text{n}} \right] $$$$ {\text{RC}}\left( {{\text{ = O}}} \right){\text{ - }}\left[ {\text{e}} \right]{ + {\rm R}^\prime {\rm NH} - }\left[ {\text{n}} \right] \to {\text{RC}}\left( {{\text{ = O}}} \right){\rm NHR^\prime} $$(Reactions joining (reactive) synthons, such as the last one, are automatically performed by gensyn, either inter- or intra-molecularly. The only other reactions applicable to such synthons are those that generate additional open valences.) Because of these explicit synthon intermediates, gensyn sequences incorporate about 50% more steps than does the same sequence represented conventionally, as carried out in the laboratory. Chemist-users seem comfortable seeing these explicit reactive intermediates.
-
The addition and removal of generic protective groups are reactions like any other. A capability to suggest specific protective groups appropriate for a user-selected reaction sequence is contemplated, based on the tables in Greene and Wuts [29].
-
In order to generate synthons appropriate for topomer searching, formally concerted two component cyclizations typically delete large numbers of atoms from one reactant and add them to the other. For example, the condensation of thioamides with α-halocarbonyls to yield thiazoles is represented by two synthons, one formed by immediately building a thiazole ring onto the α-halocarbonyl atoms, and the other by deleting the entire thioamide moiety (as exemplified by the first reaction in Fig. 3). The two open valences are labeled so that any use of one of these synthon types in a final product must also include the other. Another important aspect of cyclizations is a cost-driven requirement that one of the reactants must be symmetric, to avoid formation of regioisomers mixtures needing separation (unless as in this thiazole example the asymmetries themselves produce a single dominant regioisomer).
-
Certain synthetic complexities are currently omitted from AllChem capabilities, partially because of the economic importance of straightforward chemistry to TDR. Examples of such omissions include stereospecificity and most aromatic electrophilic substitution reactions.
-
The primary constraint on any synthetic sequence is its cumulative “cost,” roughly five AllChem-type steps (an average reaction step has a cost of −5 and the maximum sequence cost is by default −25). Other constraints on structural output include one or no prochiral atoms (so final products will not be diastereomeric) and heavy atom count (so that final libraries of products will be sufficiently “lead-like”).
-
Gensyn structure production uses identical code to operate in two modes, the Oracle-linked production mode shown in Fig. 1, and an ASCII-file based research mode capable of generating complete diagnostics for the application of specific reactions to specific structures.
Reaction description language
The starting point for gensyn’s reaction descriptions is “Sybyl Line Notation” (SLN) [30], a functionally rich notation for chemical structures based on the more widely known SMILES notation. Here is an example of a complete current description for a structurally very simple reaction, the conversion of an O–H into a nucleophilic synthon:
ID 2 O_n |
SLN HOC |
HOW MARKX,1,X1 |
VCLASS X1,n |
COST + 2 |
RXN_CLASS ActiveH 2 |
INCOMPAT N[not = N*Pr,N*Hev = Het]H |
EQUIV 1 all |
EQUIV_ORDER HOC(=O) HOC:Hev HOCH2Hev HOCH(Hev)Hev HOC(Hev)(Hev)Hev |
VRXN_CLASS 2 56,57,58,59 |
An “ID” line begins description of a new reaction, by associating a user-understandable name “O_n” with an internal identifier “2.” The “SLN” line defines the connected pattern of atoms and bonds that must be present in a structure to apply this reaction, as “HOC.” The “HOW” line describes how the synthon is to be generated, by listing the individual operations to be performed on the SLN pattern. Here there is only one such operation, the conversion of a real atom into an open valence “MARKX,” to be labeled “X1,” with the disappearing real atom being atom “1” (the H) within the SLN pattern. The “VCLASS” line defines the reactivity of “X1” as “n” (nucleophilic). A separate table records mutually reactive “VCLASS”‘s, in this case reporting that “n” reacts with “e.” The relative “COST” of this synthetic step is “+2” (which when added to the average step cost of -5 yields a total step cost of -3, for a relatively easy reaction).
In the laboratory, most reactions on most reactants are seriously complicated by the presence of other more reactive groups. The “RXN_CLASS” line classifies this reaction as a member of the “ActiveH” family. A separate list of all the SLNs, which are members of the “ActiveH” family is ordered by their descending reactivities. A prospective reactant is checked for the occurrence of any of these SLN patterns, succeeding when the “SLN” triggering this reaction is encountered but failing if any of the earlier more reactive SLN patterns is found in the reactant. The “INCOMPAT” line considers the same issue in a reaction-specific manner, by listing the SLNs of other groups whose presence prevents this specific reaction, here any “N..H” whose nitrogen is not protected (“N*Pr”) or is not amidic (“N*Hev = Het”).
The two “EQUIV” lines provide guidance when the reactant contains multiple occurrences of “SLN.” As an example in this case, consider the reactant glycerol (HOCH2CH(OH)CH2OH). The “all” keyword enables generation of the trivalent synthon “[n]-OCH2CH(O-[n])CH2O-[n],” by applying the “HOW” operation to each occurrence of “HOC.” The “EQUIV_ORDER” information guides the possibility of generating a monovalent synthon. Its SLN patterns are compared, left-to-right in presumed order of decreasing reactivity, with the three matches for “HOC” within glycerol, starting the comparison with atom “1.” The first successful comparison is with the pattern “HOCH2Hev,” which indicates that the monovalent synthon “[n]-OCH2CH(OH)CH2OH” could be a product from glycerol. It may then be objected that in glycerol there are actually two HOCH2Hev patterns, which should prevent formation of the monovalent synthon. However in this case gensyn further recognizes that those two HOCH2Hev patterns are completely identical, hence interchangeable, and so the monovalent synthon appears.
The “VRXN_CLASS” line links this generalized reaction description to more precisely defined reaction subclasses, ones that TDR chemists have previously identified as behaving distinctively. For example, “56” references primary alcohols, which are desirable to handle together in parallel laboratory synthesis because of their similarly high reactivities. The “2” references the “O” within the original “SLN.”
Searching AllChem and viewing results
The principles of topomer shape similar [31] or shape superior (3D-QSAR based) searching [32] have been described elsewhere. However, one important question, especially with so many structures, is validation of the search process. How much confidence can one have in its integrity? We have relied upon repeated “self-searches,” in which a query structure is generated by joining appropriately reactive but otherwise randomly chosen synthons. A “self-search” is successful whenever a similarity search using such a query “finds itself” at a very low-shape similarity threshold.
Viewing the results, however, presents several new challenges:
-
Allowing the user to explore costs as well as benefits, by providing access to possible synthetic routes as well as indications of favorable biology.
-
Handling the extremely numerous candidates. If only one in a trillion structures has enough biological promise to consider further, then within a database that references 1020 structures, there must be ten million hits. Or, put differently, wherever ChemSpace returned a single hit, AllChem should return a million.
Viewing synthetic routes
Because of a pressing need for assessing gensyn output, the first challenge to be addressed was viewing synthetic routes. Of course gensyn usually finds multiple synthetic routes to any particular structure. Whenever a newly produced structure is identical to one already in the database, the information about the additional route is saved into the Reactions table, and any improvement to the cumulative “cost” of synthesizing that structure replaces the previous data. Thus the information required by a synthetic route viewer is readily accessible.
The simple reaction viewer exemplified in Figs. 2 and 3 proved to be the most convenient format for users, after trials of several more complex designs. The number of structures and arrows shown is limited to what can appear in a single row. The final product of course appears at the right of such a row, with an arrow pointing to the leftmost structure in the row unless that structure is an initial building block. The row shown initially corresponds to the lowest “cost” route. The existence of other reactions producing a particular structure is signaled by a “+”just under the arrow, and double clicking on that “+” displays those other routes in a subordinate window. Double clicking on a structure generates the (lowest cost) row that ends with that structure. (Thus, Figs. 2, 3 were actually assembled from two reaction viewer displays). These figures also show how bimolecular reactions (aldol condensation and thiazole formation) are depicted.

Example of a proposed synthetic route for a novel hydroxamic acid scaffold

Example of a proposed synthetic route for a novel thiazole scaffold
Viewing and manipulating AllChem output
AllChem’s current user interface functionality does not greatly differ from that in ChemSpace. (However a need for portability and substantially improved performance resulted in a complete rewrite, including transition of the code base from Java to Python.) The user can request searches for either complete structures or individual synthons, using any combination of filters such as (topomer) shape similarity, (automatically generated topomer CoMFA) potency predictions, size, hydrophobicity, chemical reactivity, and synthetic accessibility. Shape dissimilarity filters, for example to established harbingers of undesirable off-target human side effects, could become especially important. Docking calculations can also be applied wherever desirable, once the candidates are few enough.
Before presentation to the user, AllChem search results are organized into ad hoc combinatorial libraries, grouped by query fragmentation and by open valence reactivity classes, as detailed below. These groups of similar libraries become a row in a spreadsheet, which can alternatively be viewed as a panel of (scaffold) structures. A selected spreadsheet row (or paneled structure) can be “expanded” to show the currently acceptable R groups (side chains), either individually or combined, also within a spreadsheet or structure panel. Right-clicking on any structure provides access to its synthetic routes using the above-mentioned viewer. Sorting and further filtering of the spreadsheet rows are operations critical in the translation of these ideas into laboratory syntheses.
Results and discussion
The current performance of the system will be presented from two points of view, corresponding to TDR’s two primary application priorities of lead-hopping guidance and novel scaffold ideas, and therefore involving somewhat different synthon data bases.
Leadhopping guidance
Lead hopping syntheses (seeking similar biological activities from structures that differ yet are similar in their overall shape) have usually targeted scaffold variations only, the side chains being limited to those which are readily available commercially and already “drug-compliant.” These “business rules” limit the size of AllChem’s “leadhopping guidance” database to around 250,000 synthons, not very different from the current ChemSpace database and only about 5% of the “full scale” AllChem database size. Such a smaller database is also convenient for performing summary studies that address the three following questions:
-
Does the similarity searching technology perform dependably?
-
Does shape similarity searching generate appropriate leadhop candidates?
-
Do the product structures from such searches have a sufficiently drug-like character?
To assess the reliability of similarity searching, 19 “self-searches” were performed as mentioned above. Eighteen of the 19 searches recovered the query structure perfectly, with reported overall shape differences of 0 “topomer units” for each. The 19th also recovered the query structure as the closest hit, but (because of inconsistency in nitro group valencies) with a shape difference of 40 U (from an adventitious H). Search times for these 19 self-search trials averaged 7.1 min, consistent with the search times of several hours observed for searches of the full-scale AllChem database.
Figures 4 and 5 provide an impression of the user interface and some illustrative leadhop possibilities. Figure 4 overlays two screens, the background one being the initial search results spreadsheet display, where each line corresponds to a user query (in this case one of the 19 self-searches). Double-clicking on query 5 has generated the foreground display, a spreadsheet-based index to all the search results from query 5. Each of its rows corresponds to a particular fragmentation (“Pieces” and “FragPattern” columns) and combinatorial synthesis (“R1” and “R2” columns reporting the side chain reactivities that are common to a row). The remaining columns provide summary information about the libraries referenced by a particular row. To take the highlighted row as example, the libraries that combine a nucleophilic R1, an electrophilic R2, and originate from the (3-piece) fragmentation pattern 3 include a maximum of 17 million shape-similar structures (all combinations of 210 possible cores, 8,555 possible R1’s, and 2,034 possible R2’s). The most similar structure to the query among the 17 million is shape-identical, as indicated by the values of 0 in all four MinScore cells (since the highlighted row is the one that references the successful “self-search”). Please note also how the values in the first MinScore column indicate that even the lowest shape dissimilarity within the other 22 candidate library groups shown is much higher. (When lead hopping, the usual cutoff for synthesis consideration is around 250 topomer units.)

Excerpts from the user interface for retrieving and manipulating AllChem search results. See text for details

Examples of proposed “leadhops.” The first structure is the scaffold from a query structure. The others are similar in the shapes of their topomeric alignments and are therefore as scaffolds relatively likely to confer any biological activities possessed by the query structure
Figure 5 shows some candidate scaffolds for leadhopping from a second self-search query. Since the spreadsheet row that originated Fig. 5 corresponds again to the self-search success, the most shape similar scaffold is the query fragment itself, in the upper left hand corner. All the other candidate scaffolds have rather high-shape similarities to that query fragment, although otherwise their similarities to the query fragment are rather low, and of course this is the desired result when the objective is leadhopping. Furthermore, within this synthon database all the candidate scaffolds are directly derived from building blocks that are readily available commercially (in this case aromatic/heterocyclic amines). On the other hand, the structures in Fig. 5 also call attention to an AllChem limitation that has yet to be addressed, the variability of the open valence reactivities among the synthons. The amines shown are so weakly nucleophilic that it may be quite difficult to form the urea (implied by the C(=O)-[e] moiety) especially if the other R-group (denoted by the –[n] label) is added first. Currently this issue is handled in practice by classifying synthons also by “reagent class” (see for example the brief discussion of VRXN_CLASS within the Reaction Description Language section above).
How “drug-like” are the structures representable by an AllChem (or ChemSpace) synthon collection? Figure 6 shows the distributions of molecular weight and CLOGP for the 250,000 synthons in this leadhopping—directed database. Most have molecular weights less than 350 and CLOGP values less than 4.5, as expected for a database of mostly unmodified commercial building blocks. The average molecular weights and CLOGP values for 3-synthon products would be 800 and 8.0, respectively, so that within any acceptable library the overall distribution of synthons obviously needs to be skewed well to the left of the peaks in these bar graphs. Nevertheless, ignoring the larger and more lipophilic synthons altogether would exclude any products that combine unfavorable contributions from one synthon with other synthons whose properties compensate.

Distributions of molecular weight and CLOGP values, within a synthon data base constructed by performing single steps only on commercially available building blocks
Scaffold idea generation
The other major application interest of the TDR chemists has been novel and interesting scaffold ideas. To become a component of a “lead-like” library, a scaffold must be rather small, with gensyn’s maximum number of heavy atoms for any structure typically being fourteen. Another stringent requirement, already mentioned, is no more than one prochiral center. Two relatively easy demands are a UV chromophore and a ring. The number of freely rotatable bonds should be minimal, especially where separating a diversification site from a ring. Of course there must be at least two sites of ready diversification, mostly through anticipated reactions either involving heteroatoms as nucleophiles on activated carbon electrophiles or Suzuki-type couplings. And finally the synthetic path should be short (all branches fewer than about six AllChem steps) and straightforward.
Figure 7 shows a selection of six gensyn-proposed scaffolds that meet all of these requirements. (As actually stored, the synthons have open valences that are here filled with appropriate atoms.) The first and last structures include exactly fourteen heavy atoms and none have fewer than eleven, while the prochirality limitation strongly favors unsaturation in rings, especially at fusion bonds. The proportion of gensyn-proposed synthons that are also satisfactory scaffolds usually exceeds 20% and thus requires computational filtration before inspection by the chemist, typically by diversity selection and by expanding the list of unacceptable substructures. These very approximate observations suggest that the number of accessible and structurally distinct scaffolds under these demanding constraints is on the order of 106. Just as a speculative comparison, today’s larger screening collections are of this size, but usually contain several hundred examples per scaffold, so that today’s screening collections perhaps include around 1% of the most accessible and medicinally relevant scaffold possibilities [28].

Other examples of novel scaffolds suggested by AllChem, while considering limitations on molecular weight and chirality
Figures 2 and 3 show the syntheses proposed for two further candidate cores, to give an impression of gensyn’s current strengths and weaknesses. In Fig. 2, a second point of diversification is introduced into a commercially offered cyclic hydroxamate. The first step, generic protection, seems a “strategic requirement” of the second step, but in fact gensyn is purely opportunistic, and the second step simply cannot proceed until the intermediate with the blocked OH has been generated. In performing this second step, gensyn also recognizes, first, that there are two potentially nucleophilic “ActivatedH” sites, and, second, that the two sites are equivalent so only one product is formed (marked by [n] to indicate a nucleophilic open valence). The bimolecular aldol condensation is the “lowest cost” route to the last structure on the first line, the alternatives being viewable in a separate pane by double-clicking the “+” symbol. The resulting B-ketone is considered more susceptible to hydride reduction than the amide carbonyls. Removal of the resulting active hydrogen and deprotection afford a dinucleophilic scaffold, although in actual library construction the order of these final steps would probably change.
Figure 3 proposes a thiazole scaffold having an unusual substitution geometry. The first step, thiazole condensation, illustrates the non-intuitive representation of bimolecular ring formations within AllChem, as already mentioned. Because the open valences within synthons must be acyclic, such reactions are described as ring creation on one reactant, by making two bonds to an appropriate fragment containing a third open valence, while removing the same atoms from the other leaving a complementary valence. Of course those two new valences must be specially labeled so as to react only with each other, in this case (wrongly named!) “thiadiazole_1” and “thiadiazole_2”. In the product there is only one C-H adjacent to a hetero atom so it is converted into a Suzuki-type open valence by way of a halo intermediate. The other site of derivatization is the acid provided by the initial reagent. (Note that all the descriptions of the earlier reactions along this sequence must have accepted a free carboxyl, because otherwise the sequence would not exist.)
In summary, it appears that there is no inherent barrier to the creation of a very large searchable data base of synthetically accessible synthons and their combinatorial products. Acceptable synthetic sequences can be generated in straightforward fashion, with “full scale” database re-creations requiring around a week using two standard workstations (one being the Oracle server and the other the compute engine). Complete searches based on topomer similarity seldom require more than a few hours to finish, using the same standard hardware. The initial response from the TDR chemists also encourages the belief that laboratory scientists will want to use this kind of facility directly.
The quality of the reaction descriptions and hence the synthetic sequences can always be improved. Particularly desirable would be a better scheme for handling the variable reactivities (for example nucleophilicities as mentioned above) of open valences. Of course the current numbers of reactions, reactants, and allowed steps and therefore of synthons and products could easily be increased by orders of magnitude, but we have been cautious because of the obvious impacts on searching time and output volume.
In conclusion, considering that the technology appears quite practicable, it will be interesting to begin to assess the perceived and actual value of such a system to potential user organizations.
References
Cramer RD, Soltanshahi F, Jilek R, Campbell B (2006) Abs ACS, 231st mtg: CINF 38
Todd MH (2005) Chem Soc Rev 34:247–266
Corey EJ, Wipke WT (1969) Science 166:178
Blurock ES (1990) J Chem Inf Comput Sci 30:505–510
Gelernter H, Rose JR, Chen C (1990) J Chem Inf Comput Sci 30:492–504
Helson HE, Jorgensen WL (1994) J Org Chem 59:3841–3856
Zeigarnik AV, Bruk LG, Temin ON, Likholobov VA, Maier LI (1996) Russ Chem Rev 65:117–130
Hendrickson JB (1997) Knowl Eng Rev 12:369–386
Mehta G, Barone R, Chanon M (1998) Eur J Org Chem 7:1409–1412
Matyska L, Koca J (1991) J Chem Inf Comput Sci 31:380–386
Young DC (2002) in Comp. Chem. Wiley, NYC, NY
Lushnikov DE, Zefirov NS (1992) J Chem Inf Comput Sci 32:317–322
Johnson AP, Marshall C, Judson PN (1992) J Chem Inf Comput Sci 32:411–417
Bertz SH, Rucker C, Rucker G, Sommer TJ (2003) Eur J Org Chem 24:4737–4740
Satoh H, Funatsu K (1995) J Chem Inf Comput Sci 35:34–44
Moll H (1994) J Chem Inf Comput Sci 34:117–119
Hanessian S, Franco J, Gagnon G, Laramee D, LaRouche B (1990) J Chem Inf Comput Sci 30:413–425
Ihlenfeldt W-D, Gasteiger J (1995) Angew Chem Intl 34:2613–2633
Caflisch A, Karplus M (1995) Pers Drug Disc Des 3:51–84
Jauffret P, Ostermann C, Kaufmann G (2003) Eur J Org Chem 10:1983–1992
Baber JC, Feher M (2004) Mini Rev Med Chem 4:681–692
Ugi I, Bauer J, Bley K, Dengler A, Dietz A, Fontain E, Gruber B, Herges R, Knauer M, Reitsam K, Stein N (1993) Angew Chem Intl Ed 32:201–227
Jilek RJ, Cramer RD (2004) J Chem Inf Comp Sci 44:1221–1227
Cramer RD, Jilek RJ, Guessregen S, Clark SJ, Wendt B, Clark RD (2004) J Med Chem 47:6777–6791
Cramer RD, Patterson DE, Clark RD, Soltanshahi F, Lawless MS (1998) J Chem Inf Comp Sci 6:1010–1023
Andrews KM, Cramer RD (2000) J Med Chem 43:1723–1740
Corey EJ, Cramer RD, Howe WJ (1972) J Am Chem Soc 94:440–459
Ertl P, Jelfs S, Muhlbacher J, Schuffenhauer A, Selzer P (2006) J Med Chem 49:4568–4573
Greene TW, Wuts PGM (1999) Protective groups in organic synthesis. Wiley, NYC, NY
Ash S, Cline MA, Homer RW, Hurst T, Smith GB (1997) J Chem Inf Comput Sci 37:71–79
Cramer RD, Clark RD, Patterson DE, Ferguson AM (1996) J Med Chem 39:3060–3069
Cramer RD (2003) J Med Chem 46:374–389
Acknowledgment
We are grateful to NIH/GMS for financial support of this development project, currently from award: SBIR 2 R44 GM068359-02.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cramer, R.D., Soltanshahi, F., Jilek, R. et al. AllChem: generating and searching 1020 synthetically accessible structures. J Comput Aided Mol Des 21, 341–350 (2007). https://doi.org/10.1007/s10822-006-9093-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-006-9093-8