
Abstract
Purpose
We aim to investigate whether there is a genetic predisposition in women who developed ovarian hyperstimulation syndrome (OHSS) after GnRH antagonist protocol with GnRH agonist trigger and freeze-all approach.
Methods
Four patients with OHSS after GnRH agonist trigger and freeze-all approach were gathered from the worldwide patient population. These patients were analyzed through Whole Exome Sequencing. In this study known causes of OHSS were investigated and new causes present in at least two individuals were searched for.
Results
In the first part of the study, we evaluated the presence of mutations in genes already known to be involved in OHSS. In PGR and TP53, heterozygous alterations were detected. PGR is predicted to be involved in progesterone resistance with a recessive inheritance pattern and is, therefore, not considered as being causal. The consequences of the variant detected in TP53 currently remain unknown. In part 2 of the study, we assessed the clinical significance of variants in genes previously not linked to OHSS. We especially focused on genes with variants present in ≥ 2 patients. Two patients have variants in the FLT4 gene. Mutations in this gene are linked to hereditary lymphedema, but no link to OHSS has been described.
Conclusions
Defining a genetic predisposition for OHSS is essential in view of prevention. In this study, a potential link between the FLT4 gene and OHSS has been suggested. Future functional studies are essential to define a more precise involvement of the detected variants in the development of OHSS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ovarian hyperstimulation syndrome (OHSS) is an exaggerated response to ovarian stimulation, characterized by cystic enlargement of the ovaries, abdominal distention and pain, and fluid shift from the intravascular space to the third space, which may eventually result in ascites, pericardial and pleural effusions, and even in generalized edema. This may lead to hypovolemia, hemoconcentration, electrolyte imbalances and coagulation disorders, and even life-threatening complications such as hemorrhage from the rupture of an ovarian cyst, adult respiratory distress syndrome, thromboembolism, and acute renal failure [1].
Hypersensitivity to ovarian stimulation with exogenous gonadotropins is the most common cause of OHSS. In a subset of infertile patients, ovarian stimulation induces growth of a large number of follicles and results in high estradiol levels. Eventually, these patients are exposed to a bolus of human chorionic gonadotropin (hCG) to finalize oocyte maturation. As hCG has a longer half-life than the endogenous luteinizing hormone (LH), sustained luteotropic activity will ultimately cause abnormal vascular permeability with extravasation of fluid to the third space and, consequently, the clinical presentation of OHSS [2, 3].
The key molecules responsible for the high vascular permeability are vascular endothelial growth factor (VEGF) and factors involved in the ovarian renin-angiotensin system [4]. VEGF is produced by the granulosa cells after stimulation with gonadotropins, and its production increases substantially after the administration of hCG [5].
The VEGF systems includes three receptors (FLT1 (= VEGFR1), KDR (= VEGFR2), and FLT4 (= VEGFR3)) and six ligand molecules (VEGFA, VEGFB, VEGFC, VEGFD (= FIGF), PDGFD (= VEGFE), and PIGF). Multiple ligands can bind to one receptor: VEGA/B and PIGF can bind to VEGRF1, VEGFA/C/D/E can bind to VEGFR2, and VEGFC/D can bind to VEGFR3. Only VEGFR2 and VEGFR3 (official name FLT4) have been associated with a genetic disorder so far. Dominant pathogenic variants are associated with hereditary hemangioma [6]. So far, > 85 variants have been described in the FLT4 gene, most of them located between exon 17 and exon 28.
OHSS can occur in up to one third of all cases of high-risk patients, i.e., women with polycystic ovaries, with estradiol levels of > 3000 pg/ml on the day of hCG administration, or with 13 or more follicles with a mean diameter of at least 11 mm [7].
The risk of OHSS can be reduced by eliminating the injection of hCG as a final oocyte maturation trigger. This option is only valid when the ovaries are stimulated using a gonadotropin-releasing hormone (GnRH) antagonist protocol instead of a GnRH agonist protocol. Since GnRH antagonists inhibit pituitary function directly, the receptors can recover much faster, which is in contrast to the effect of desensitization caused by GnRH agonists. This allows clinicians to use GnRH agonists as a trigger instead of hCG, which results in a temporary displacement of the GnRH antagonists followed by an endogenous LH surge. Since a few years, the use of the latter protocol has emerged, and this has appeared to result in a dramatic reduction of the incidence of OHSS [8]. However, still at least seven cases of severe OHSS have been reported with this so-called freeze-all approach [9,10,11,12,13] . These observations suggest that other crucial components, besides sustained hCG activity, are involved in the development of OHSS. Furthermore, several cases of familial spontaneous OHSS have been described [14]. In these cases, OHSS occurred after a spontaneous pregnancy, i.e., without controlled ovarian stimulation. In both spontaneous and iatrogenic OHSS, hCG plays as a key factor to cause this syndrome. However, the fact that OHSS can also occur in women following a spontaneous pregnancy raises suspicion of a genetic component. Such a genetic trait can also be considered in women who developed OHSS despite the absence of hCG administration.
So far, the focus in the search for a genetic predisposition for OHSS was confined to known and frequent polymorphisms in OHSS-predisposing genes (Table 1). These studies are summarized in Altmäe et al. [15], Lledo et al. [16], Boudjenah et al. [17, 18], Moron et al. [19], O’Brien et al. [20], and Rizk [21]. Although these genes could be implicated in the genesis of OHSS, the presence of single nucleotide polymorphisms (SNPs) with high population frequencies, is unlikely to be involved in OHSS following treatment with the GnRH antagonist protocol and GnRH agonist trigger, or in spontaneous OHSS.
In the present study, whole exome sequencing (WES) was performed in four patients who developed OHSS despite application of a reproductive treatment protocol without the use of hCG, in order to attempt to identify potential molecular risk factors for OHSS.
Material and methods
Study design and subjects
DNA samples of three women described by Gurbuz et al. [11] and the woman previously detected by Santos-Ribeiro et al. [13] were collected for this study. These patients were treated with a GnRH antagonist protocol followed by a GnRH agonist trigger and a freeze-all approach. All subjects signed a written informed consent. Approval for the study was received from the Ethics Committee of the UZ Brussel (EC2916/67). The DNA of patient 1 was extracted from peripheral blood according the standard procedures of the Center for Medical Genetics of UZ Brussel, i.e., using the CMG-1074 kit on the Janus G3 machine (Perkin Elmer). This patient is from the Caucasian origin. Meanwhile, DNA samples from peripheral blood from patients 2, 3, and 4 were kindly provided by Dr. Gurbuz, and these patients were of Turkish origin. All patients had suffered OHSS grade 4 according to the Golan criteria (see Table 2).
Exome sequencing and data analysis
Whole exome sequencing (WES) was performed in the Centre of Medical Genetics, UZ Brussel, in collaboration with the Brussels Interuniversity Genomics High Throughput core (BRIGHT core) according to the standard procedures. First, DNA was fragmented into fragments of on average 250 bp (Covaris M220). Libraries were created through the KAPA Hyper Prep Kit and quality-controlled (AATI Fragment Analyzer and Life Technologies Qubit 2.0). Subsequently, target enrichment was performed using the SeqCap EZ v3.0 kit (Roche Diagnostics Belgium). The resulting libraries were diluted to 10 pM, followed by clonal amplification on the Illumina cBot using the TruSeq PE Cluster Kit v4-cBot-HS kit. Paired-end sequencing (2 × 125 bp) was performed on the Illumina HiSeq 1500 using the TruSeq SBS kit v4-HS (250 cycles) in order to obtain a 75× minimum average coverage.
Data analysis
Raw WES data were quality-controlled by using FastQC and mapped to the human reference genome with BWA (0.7.10). Mapping qualities were assessed via overall coverage analysis by an in-house designed script. The mapped reads were processed using the GATK (2.7.2) pipeline (IndelRealaginer, BaseRecalibrator, HaplotypeCaller), and the detected variants were annotated by Alamut Batch.
Further selection and filtering of variants was done with the use of Highlander, an in-house developed software algorithm for variant classification (Université Catholique de Louvain, Belgium). Variants were filtered over several variant databases (dbSNP, ExAC) using tools predicting splice effects (MaxEntScan, SpliceSiteFinder). Synonymous variants not effecting splicing and those with an allele frequency > 2% (dbSNP, GnomAD) were excluded.
From this pool of variants, a first selection was performed at the gene level, i.e., genes that had previously been studied in view of OHSS (Table 1). Next, using Highlander, genes were selected in which variants were detected in at least two patients. From this selection, a manual curation was performed, as described in the “Results” section.
Sanger sequencing
The presence of the detected variants in the FLT4 gene was confirmed by the Sanger sequencing. For this, PCR amplification with 250 ng of DNA was performed with a touchdown PCR according to standard procedures. Primers for the analysis were M13 tagged and developed by IDT (TGCTCGACTGCAAGAACGTG and CTGCACTTAGCAGGAGGACC for exon 13, CGCCACCCAGCCTTCTTCTC and TGCCACCAGAGTTCAACCAG for exon 30, RefSeq NM_182925.4). The Sanger sequencing was also performed with M13 primers, according to standard procedures.
Results
Based on the available literature, we decided to initially study the genes that have been previously associated with OHSS (Table 1). A single variant in the TP53 gene (NM_000546.4) was detected in patient 1. It is a variant of unknown significance: c.868C>T, p.(Arg290Cys). This substitution is located at a moderately conserved nucleotide and amino acid substitution. The population frequency is very low, being 0.001% (or 4/277216 genomes). Prediction programs SIFT, PolyPhen2, and MutationTaster predict that this variant is pathogenic.
A second variant was detected in the PGR gene (NM_000926.4) of patient 2: c.97G>T, p.(Ala33Ser). This novel variant is not conserved at the nucleotide level and only weakly conserved at the amino acid level. There is a moderate physicochemical difference between alanine and serine. Consequently, all in silico prediction programs point towards a neutral variant.
In the second part of the study, genes previously not linked to OHSS were investigated. Since all individuals have multiple rare variants, we looked at genes with one or more variants in at least two of our patients and a population frequency < 2%. A total of 108 genes fulfilled these criteria. These (108) genes were then investigated in more depth. First, genes with at least two variants or homozygous variants (i.e., variants associated with potential recessive disorders) were investigated. Following thorough literature review to assess the function of these genes, no candidate genes remained. Therefore, it was acceptable to further decrease the cut-off for the population frequency to 1%, generally accepted for rare dominant disorders. A total of 25 genes could be removed from the selection. The remaining (variants in) 83 genes were evaluated individually: variants with a population frequency > 1% in one of the ethnical sub-populations in the GnomAD database and genes associated with a known disease not linked to OHSS, were removed.
Finally, 38 genes remained in our selection. These are mostly genes with an unknown function. Among these genes, four genes (KIF26A, PTPN13, TBC1D2, and ZNF500) were present in which variants were detected in three patients. In ZNF500 (NM_021646.3), the same variant was detected in three patients (patients 2, 3, and 4): c.461G>A, p.(Gly154Glu). This variant has a general population frequency of 0.07% and is the most frequent in the South-Asian population (0.2%). All three patients were of the same origin, and consequently, this might constitute a founder effect. This variant is not conserved and presumably represents a SNP according to different prediction programs. All other variants were different in each patient.
In the final selection of 38 genes, one gene that caught our attention was the FLT4 gene (NM_182925.4). This was the only gene remaining in our selection, linked to a known disorder: pathogenic variants in this gene cause an autosomal dominant hereditary lymphedema IA. In two women, a variant was detected in this gene (patients 2 and 3): c.1985A>T, p.(Asp662Val) and c.3908G>C, p.(Gly1303Ala). The first variant has never been detected before. This variant is located in the immunoglobulin-like domain. The second variant was present in multiple populations with an overall frequency of 0.3%. The highest population frequency was detected in a South-Asian population where it is present in 1% of the population. This variant has been reported by Mattassi et al. [22] in a patient with vascular anomalies. The first variant is pathogenic according to MutationTaster; the second variant should be classified as a polymorphism according to different prediction programs tested (AlignGVGD, SIFT, MutationTaster, and PolyPhen2).
Discussion
The aim of this study was to explore rare genetic variants potentially involved in OHSS. By selecting variants present in < 2% of the general population, frequent variants/polymorphisms (e.g., FSHR or MTHFR) in genes potentially linked to OHSS (mentioned in Table 1) were filtered out. As these variants are very common, it is unlikely that they are indeed causal for the development of OHSS with the present treatment protocol. However, it cannot completely be ruled out that they may “slightly” impair the phenotype.
In one of the genes in which polymorphisms have been associated with OHSS, a rare variant was detected: TP53, c.868C>T, p.(Arg290Cys). This nucleotide alteration has not been reported previously as being pathogenic, although amino acid alterations at the same position (p.(Arg290His) and p.(Arg290Leu)) have been associated with Li-Fraumeni syndrome, a hereditary cancer predisposition syndrome.
So far, patient 1 has not developed any symptoms compatible with a diagnosis of Li-Fraumeni syndrome. Yet, the presence of this variant of unknown significance might have important consequences for the patient and her family. Therefore, a surveillance program should be considered in this family. A previously reported polymorphism p.(Pro72Arg) in the TP53 gene (coding for the P53 protein) has been associated with recurrent embryo implantation failure. This patient and two other patients included in this study are heterozygous for this SNP. However, given the high population frequencies (up to 74%, Boudjenah et al. [18]), the presence of this SNP will likely have no functional consequences.
Overall, it is unknown whether TP53 and more specifically the c.868C>T, p.(Arg290Cys) variant is associated with OHSS. Therefore, this patient was not excluded from part two of the study.
In the first part of the study, looking at genes previously linked to OHSS (Table 1), one more variant was detected in a gene coding for the progesterone receptor. Mutations in PGR are possibly linked to progesterone resistance, with an autosomal recessive inheritance pattern. Since only a single variant was detected in our patient, which is likely a neutral variant according to different prediction programs, this variant is probably not involved in OHSS.
In a second part of the study, we investigated genes that had not yet been associated with OHSS. Of the initial 108 candidate genes in which SNPs were detected in at least two patients, a further reduction to 38 genes was performed. Of these, 4 genes were present in three out of four patients. In ZNF500, the same variant was detected in three patients. These were three patients from Turkish origin. Consequently, this might be a variant linked to this subpopulation. According to our in-house database, this variant was detected only in one man with cardiac problems from Caucasian origin. Little is known about this gene, except that it is expressed in multiple tissues [23].
The remaining genes in which variants are detected in three patients were KIF26A, PTPN13, and TBC1D2. Although all three genes are expressed at low levels in ovaries, they are unlikely to have a major function in this organ.
One gene of interest was the FLT4 gene. The FLT4 gene is coding for the VEGFR3 protein, a protein involved in the regulation of lymphatic vessel function. The family of VEGF proteins and their receptors (VEGFR) are known to play a key role in the development of OHSS (reviewed in Soares et al. [24]), particularly VEGFR2 and the VEGF(A) ligand. Binding of VEGF(A) to its receptor is associated with an increased vascular permeability. Moreover, it has been shown that gonadotrophins as well as hCG largely increase the expression of Vegf and Vegfr2 in vivo (rat models) [25]. The abnormal increase of VEGF and VEGFR2 production promotes the development of OHSS. Multiple studies have also shown that the levels of VEGFA correlate to the risk of developing OHSS in humans (Soares et al. [24]).
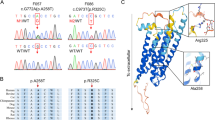
In the present study, we detected two variants in the FLT4 gene. Mutations in FLT4 (coding for VEGFR3) cause autosomal dominant hereditary lymphedema IA (Milroy’s disease) characterized by the presence of lymphedema, predominantly in the lower limbs. To our knowledge, the two patients do not have major symptoms of Milroy’s disease. However, the expression of this disease is highly variable. That said, the detected variants are mild and do not have a major impact on the disease. The function of VEGFR3 in the development of OHSS remains less understood. However, it is known that VEGFR3 interacts with VEGFR2 to form homo- and heterodimers (reviewed in Bahram et al. [26]) (Fig. 1). The regulation of the interaction between these proteins is crucial for their correct function and is tissue-dependent. Heinolainen et al. [27] also showed that Vegfr3 regulates Vegfr2 expression and the Vegf/Vegfr2 pathway activity in mice. When there is a deletion of Vegfr3, vascular permeability is increased due to a lack of inhibition of the VEGF/VEGFR2 pathway. The authors suggested that the VEGFR3 protein inhibits Vegfr2 mRNA transcription. Mäkinen et al. [28] has shown that blocking of VEGFC protein by adding soluble VEGFR3 (i.e., only the ligand binding domain) causes lymphedema. Here, downstream signaling is blocked by the lack of the internal domain [28].

Interaction between VEGFR2 and VEGFR3 in normal (1A) and pathologic conditions (1B). The potential consequences for the detected FLT4 variants are either (1) the binding of VEGFC (or VEGD) is less efficient with more VEGFC (or VEGD) remaining available for binding to VEGFR2 or and (2) internalization of VEGFR3 is hampered which may impair the negative feedback regulation of VEGFR2, increasing the expression or VEGFR2
Alternatively, there may be a competition in the binding of ligands to VEGFR2 and VEGFR3 receptors (Fig. 1). The ligands for VEGFR3 are VEGFC and VEGFD. These ligands also bind to VEGFR2, together with VEGFA and VEGFE. By blocking VEGFC (and potentially also VEGFD), competition between the ligands for binding to VEGFR2 is missing, and more VEGFA substrate will be able to bind VEGFR2.
So far, most naturally occurring mutations in FLT4 causing Milroy’s disease are located between exon 17 and exon 28, in the protein kinase domain, a domain essential for downstream signaling. It remains unknown whether patients with mutations causing Milroy’s disease are more prone to develop OHSS. The alterations we detected are located in exon 13 and exon 30. The first one is located in the immunoglobulin-like domain, responsible for binding of the ligands. Consequently, the detected variant might influence the binding of its ligands (VEGFC or VEGFD), thereby causing preferential binding to VEGFR2 homodimers instead of VEGFR3 homo/heterodimers. The second alteration is located in the C-terminal region. The role of this C-terminal region remains unknown. For VEGFR2, this region is important for internalization of the receptor and consequently for the function of the receptor. Possibly, the detected variant in the C-terminal region is important for internalization of VEGFR3, which will influence the expression of VEGFR3 and the binding to VEGFR2. Alternatively, the regulation of VEGFR2 mRNA expression may be altered.
Conclusion
This is the first study linking VEGFR3 (or the FLT4 gene) to OHSS. However, only four cases were included in this study, among which two patients had variants in the FLT4 gene. Since only seven patients worldwide have been described with severe OHSS in a GnRH antagonist protocol with GnRH agonist trigger and cryopreservation of all embryos, it will be hard to confirm these data. Consequently, functional studies are essential to identify the true role of the VEGFR3 receptor in OHSS. Furthermore, the impact of the variants detected in the present study remains to be elucidated. This study shows that a subset of patients who develop OHSS might have an underlying genetic defect. However, since OHSS has become not only a rare but also largely unpredictable condition, the question could be raised whether women who undergo a fertility treatment involving ovarian stimulation should be screened for the presence of variants in OHSS risk genes. The cost-efficiency ratio of such a preliminary screening step may have to be considered. In the meantime, it remains mandatory to use ovarian stimulation protocols adapted to the OHSS risk profile of the patient, combined with intensified monitoring and surveillance of the patient.
References
Delvigne A, Rozenberg S. Review of clinical course and treatment of ovarian hyperstimulation syndrome (OHSS). Hum Reprod Update. 2003;9:77–96.
Soares SR, Amols MH, Hudson SBA, et al. Etiology of OHSS and use of dopamine agonists. Fertil Steril. 2012;97:517–22.
Fatemi HM, Garcia-Velasco J. Avoiding ovarian hyperstimulation syndrome with the use of gonadotropin-releasing hormone agonist trigger. Fertil Steril. 2015;103:870–3.
Rizk B, Aboulghar M, Smitz J, Ron-El R. The role of vascular endothelial growth factor and interleukins in the pathogenesis of severe ovarian hyperstimulation syndrome. Hum Reprod Update. 1997;3:255–66.
Neulen J, Yan Z, Raczek S, Weindel K, Keck C, Weich HA, et al. Human chorionic gonadotropin-dependent expression of vascular endothelial growth factor/vascular permeability factor in human granulosa cells: importance in ovarian hyperstimulation syndrome. J Clin Endocrinol Metab. 1995;80:1967–71.
Walter JW, North PE, Waner M, Mizeracki A, Blei F, Walker JWT, et al. Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer. 2002;33:295–303.
Delvigne A. Epidemiology of OHSS. Reprod BioMed Online. 2009;19:8–13.
Berkkanoglu M, Coetzee K, Bulut H, Ozgur K. Risk of ovarian torsion is reduced in GnRH agonist triggered freeze-all cycles: a retrospective cohort study. J Obstet Gynaecol. 2018:1–6. https://doi.org/10.1080/01443615.2018.1479381.
Griesinger G, Schultz L, Bauer T, Broessner A, Frambach T, Kissler S. Ovarian hyperstimulation syndrome prevention by gonadotropin-releasing hormone agonist triggering of final oocyte maturation in a gonadotropin-releasing hormone antagonist protocol in combination with a “freeze-all” strategy: a prospective multicentric study. Fertil Steril. 2011;95:2029–33.
Fatemi HM, Popovic-Todorovic B, Humaidan P, Kol S, Banker M, Devroey P, et al. Severe ovarian hyperstimulation syndrome after gonadotropin-releasing hormone (GnRH) agonist trigger and “freeze-all” approach in GnRH antagonist protocol. Fertil Steril. 2014;101:1008–11.
Gurbuz AS, Gode F, Ozcimen N, Isik AZ. Gonadotrophin-releasing hormone agonist trigger and freeze-all strategy does not prevent severe ovarian hyperstimulation syndrome: a report of three cases. Reprod BioMed Online. 2014;29:541–4.
Ling S-Y, Chong K-M, Hwang J-L. Persistent megalocystic ovary following in vitro fertilization in a postpartum patient with polycystic ovarian syndrome. Taiwan J Obstet Gynecol. 2006;45:70–2.
Santos-Ribeiro S, Polyzos NP, Stouffs K, de Vos M, Seneca S, Tournaye H, et al. Ovarian hyperstimulation syndrome after gonadotropin-releasing hormone agonist triggering and “freeze-all”: in-depth analysis of genetic predisposition. J Assist Reprod Genet. 2015;32:1063–8.
Montanelli L, Delbaere A, Di Carlo C, et al. A mutation in the follicle-stimulating hormone receptor as a cause of familial spontaneous ovarian hyperstimulation syndrome. J Clin Endocrinol Metab. 2004;89:1255–8.
Altmäe S, Hovatta O, Stavreus-Evers A, Salumets A. Genetic predictors of controlled ovarian hyperstimulation: where do we stand today? Hum Reprod Update. 2011;17:813–28.
Lledo B, Ortiz JA, Llacer J, Bernabeu R. Pharmacogenetics of ovarian response. Pharmacogenomics. 2014;15:885–93.
Boudjenah R, Molina-Gomes D, Torre A, Boitrelle F, Taieb S, Dos Santos E, et al. Associations between individual and combined polymorphisms of the TNF and VEGF genes and the embryo implantation rate in patients undergoing in vitro fertilization (IVF) programs. PLoS One. 2014;9:e108287.
Boudjenah R, Molina-Gomes D, Torre A, Bergere M, Bailly M, Boitrelle F, et al. Genetic polymorphisms influence the ovarian response to rFSH stimulation in patients undergoing in vitro fertilization programs with ICSI. PLoS One. 2012;7:e38700.
Morón FJ, de Castro F, Royo JL, Montoro L, Mira E, Sáez ME, et al. Bone morphogenetic protein 15 (BMP15) alleles predict over-response to recombinant follicle stimulation hormone and iatrogenic ovarian hyperstimulation syndrome (OHSS). Pharmacogenet Genomics. 2006;16:485–95.
O’Brien TJ, Kalmin MM, Harralson AF, Clark AM, Gindoff I, Simmens SJ, et al. Association between the luteinizing hormone/chorionic gonadotropin receptor (LHCGR) rs4073366 polymorphism and ovarian hyperstimulation syndrome during controlled ovarian hyperstimulation. Reprod Biol Endocrinol. 2013;11:71.
Rizk B. Symposium: Update on prediction and management of OHSS. Genetics of ovarian hyperstimulation syndrome. Reprod Biomed Online. 2009;19:14–27.
Mattassi R, Manara E, Colombo PG, Manara S, Porcella A, Bruno G, et al. Variant discovery in patients with Mendelian vascular anomalies by next-generation sequencing and their use in patient clinical management. J Vasc Surg. 2018;67:922–932.e11.
Hsu C-C, Chiang C-W, Cheng H-C, Chang WT, Chou CY, Tsai HW, et al. Identifying LRRC16B as an oncofetal gene with transforming enhancing capability using a combined bioinformatics and experimental approach. Oncogene. 2011;30:654–67.
Soares SR, Gómez R, Simón C, García-Velasco JA, Pellicer A. Targeting the vascular endothelial growth factor system to prevent ovarian hyperstimulation syndrome. Hum Reprod Update. 2008;14:321–33.
Gómez R, Simón C, Remohí J, Pellicer A. Administration of moderate and high doses of gonadotropins to female rats increases ovarian vascular endothelial growth factor (VEGF) and VEGF receptor-2 expression that is associated to vascular hyperpermeability1. Biol Reprod. 2003;68:2164–71.
Bahram F, Claesson-Welsh L. VEGF-mediated signal transduction in lymphatic endothelial cells. Pathophysiol Off J Int Soc Pathophysiol. 2010;17:253–61.
Heinolainen K, Karaman S, D'Amico G, Tammela T, Sormunen R, Eklund L, et al. VEGFR3 modulates vascular permeability by controlling VEGF/VEGFR2 signaling. Circ Res. 2017;120:1414–25.
Mäkinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001;7:199–205.
Acknowledgments
We wish to thank the lab technicians of the Center for Medical Genetics for performing WES and Sanger sequencing. Funding was received from ‘Wetenschappelijk Fonds Willy Gepts of the UZ Brussel’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Approval for the study was received from the Ethics Committee of the UZ Brussel (EC2916/67).
Rights and permissions
About this article

Cite this article
Stouffs, K., Daelemans, S., Santos-Ribeiro, S. et al. Rare genetic variants potentially involved in ovarian hyperstimulation syndrome. J Assist Reprod Genet 36, 491–497 (2019). https://doi.org/10.1007/s10815-018-1372-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-018-1372-5