
Abstract
Benign hydatidiform mole, complete or partial, is the most common type of gestational trophoblastic disease (GTD) characterised by excessive trophoblastic proliferation and abnormal embryonic development. Although most complete hydatidiform moles (CHMs) are diploid androgenetic, a few cases of CHMs are biparental, characterised by recurrence and familial clustering. In these rare cases, mutations in NLRP7 or KHDC3L genes, associated with maternal imprinting defects, have been implicated. Current data regarding future pregnancy options in hydatidiform moles are discussed and our opinion is presented based on an incidence that took place in our hospital with a woman with consecutive molar pregnancies. In recurrent hydatidiform moles, DNA testing should be performed and when NLRP7 or KHDC3L mutation are detected, oocyte donation should be proposed as an option to maximise woman’s chances of having a normal pregnancy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gestational trophoblastic disease (GTD) is a spectrum of cellular proliferations arising from the placental villous trophoblast encompassing four main clinopathologic forms: hydatidiform mole (complete or partial), invasive mole, choriocarcinoma and placental site trophoblastic tumour [1]. The incidence of GTD varies between different countries with the highest rates found in parts of Asia. Approximately 15% of CHMs and 5% of PHMs transform into one of the malignant forms of GTD [2], thus it is of great importance to accurately diagnose the disease. Benign hydatidiform mole (HM), complete or partial, is the most common type of GTD. It is a human pregnancy characterised by excessive trophoblastic proliferation and abnormal embryonic development and it can be sporadic or recurrent. Recurrent hydatidiform moles (RHMs) are defined by the occurrence of two or more molar pregnancies in the same patient. RHMs may be sporadic, occurring in a single individual in a family or may be familial as in biparental moles (BiCHM) that have both a maternal and a paternal contribution and are due to an autosomal recessive defect in the female germ line [3]. In women who have at least two episodes of molar pregnancy, assisted reproductive technology may help to achieve normal fertilisation of oocytes. However, even embryos that come from standard in vitro fertilisation (IVF) techniques can lead to HM and failure to achieve normal pregnancy. We present our opinion and current data regarding future pregnancy options in hydatidiform moles with respect to an incidence that took place in our hospital with a woman with consecutive molar pregnancies.
Molar pregnancy: a complex genetic game of chess
Hydatidiform mole is an abnormal pregnancy, characterised by placental overgrowth, while the embryonic development is severely abnormal or absent. Histopathologically, it can be classified as complete or partial. The placenta of both CHM and PHM is characterised by edematous swelling of the chorionic villi and trophoblastic hyperplasia (Fig. 1). The incidence of CHM is around 1/1000 pregnancies and of PHMs around 3/1000 [1]. Oocytes from older women and teenagers are at increased risk of molar gestation [4]. After a molar pregnancy, the risk of an additional, complete and partial, mole rises to 1–2%, whereas after two molar pregnancies, the risk of a third mole is 15–20%, which is not decreased by changing of the partner [5]. Furthermore, these women are more likely to develop persistent gestational trophoblastic tumour [6].

Complete hydatidiform mole of our case (haematoxylin-eosin stain, magnification × 100) (villus edema, hyperplasia of trophoplastic tissue, mild nuclear atypia)
CHM and PHM are genetically different in that CHM has a normal, diploid number of chromosomes, while PHM is triploid. Most CHMs result from fertilisation of an apparently empty ovum (null nuclear genome) by a haploid sperm which then duplicates its own chromosomes, restoring the diploid chromosome number (homozygous monospermic mole) [6]. The chromosome constitution of these moles is usually 46,XX (46,YY has never been observed and thus probably non-viable). Approximately, 20–25% of CHMs result from fertilisation of an empty ovum by two sperms (dispermic or heterozygous mole) and their chromosome constitution is 46,XX or 46,XY. How an unnucleated oocyte is produced is unclear; however, one possible error could be a non-disjunction of all chromosomes during meiosis, with all chromosomes ending up in one of the polar bodies. CHMs show no evidence of fetal tissues. Tetraploid and even triploid CHMs are possible, as long as all the extra copies of the chromosomal sets are paternally derived [4]. Except for the androgenetic CHMs (only paternal contribution to the genome), occasional diploid CHMs that have both a maternal and paternal contribution to the genome also exist. These biparental CHMs (BiCHM), although rare, are of great interest as they have been associated with recurrent familial molar pregnancies.
Regarding PHMs, they generally result from the fertilisation of an apparently normal haploid ovum by two spermatozoa [7], although occasional PHMs have been reported to come from fertilisation of an ovum by a diploid sperm. Thus, PHMs are triploid in origin, comprising of one set of maternal haploid chromosomes and two sets of paternal haploid chromosomes, and in contrast to CHMs show evidence of fetal tissues. Rare tetraploid PHMs have also been reported with three haploid paternal chromosomal sets (92XXXX, 92XXYY, 92XXXY) [4]. In sum, most CHMs have two paternal contributions to the genome, whereas PHMs have both maternal and paternal contribution to the nuclear genome. However, occasionally diploid CHMs that have both a maternal and paternal contribution to the genome have been described and these biparental CHMs (BiCHM), although rare are mostly associated with familial recurrent molar pregnancies (Tables 1 and 2).
Current data and our experience
Familial HM was first described in three families in which trophoblastic disease occurred in one or more pregnancies in two or more sisters [8]. Subsequently, two Italian kindreds each with two affected sisters were described [9, 10], as well as a large consanguineous Lebanese family, in which two sisters and a cousin were affected [11]. Linkage and homozygosity mapping of the family identified a homozygous region on 19q13.3–13.4 segregating with the disorder [12]. This region was also implicated in a German family of three affected sisters with recurrent CHM [12, 13], as well as in other families with recurrent CHM [14, 15]. Today, more than 30 families with RHMs have been reported [4].
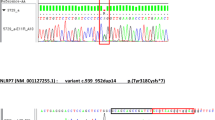
Our experience is about a 30-year-old woman of Greek origin, with a history of two previous molar pregnancies. Histopathologic examination confirmed the diagnosis of CHM (Fig. 1). In addition, one out of her four sisters had also faced the same condition in her pregnancies, having experienced two molar pregnancies. There was no consanguinity in their family. The pedigree chart of this family is depicted in Fig. 2. Genetic analyses with human whole exome sequence capture and next-generation sequencing, as well as bi-directional fluorescent automated DNA Sanger sequencing, revealed that the proband woman was homozygous for the pathogenic c.2471 + 1G > A (rs104895505) (p.Leu825X) mutation of the NLRP7 gene which has been associated with familial RHMs (Fig. 3).

Pedigree structure of our case report (black rounds cases with complete hydatidiform moles, the proband woman, homozygous for the pathogenic c.2471 + 1G > A, is indicated by the arrow

Genetic analysis of our case with the pathogenic c.2471 + 1G > A (rs104895505) mutation of the NLRP7 gene. The mutation is indicated by the arrow
Disentangling the mystery of genetic imprinting and BiCHM
Pathologically, most pregnancies in families with recurrent HMs are CHMs and specifically BiCHMs, having both a maternal and paternal genetic origin. We confirm the occasionally familial recurrent HMs have been described as PHMs. BiCHMs are due to an autosomal recessive defect in the female germ line, whereas the paternal genotype does not contribute to the pathogenesis [16]. Affected women can be identified by genotyping of the CHM. Sporadic CHMs are androgenetic, while those associated with recurrent familial HMs are diploid, biparental in origin. Acquisition of global imprinting alteration at a critical time point in a preimplantation embryo occurs through the de novo absence of the maternal haploid genome in sporadic androgenetic CHMs or through mutations of NLRP7 or KHDC3L, shutting down the entire maternal imprinting gene expression, in familial BiCHMs [4].
NLRP7, a nucleotide oligomerisation domain (NOD)-like receptor, pyrin containing 7, maps to 19q13.4 and is the first identified causative gene for RHMs. Studies from various groups and populations concur that NLRP7 is a major gene for this condition and is mutated in 48–80% of patients with at least two HMs, depending on patients’ ascertainment criteria and populations [17, 18]. To date, 47 mutations in NLRP7 have been reported in patients with two defective alleles, including stop codons, small deletions or insertions (less than 20 bp), splice mutations, large deletions or insertions, and complex rearrangements [19]. In addition to these mutations, two protein-truncating mutations, a stop codon, L823X, and a deletion of 60-kb extending from intron 8 of NLRP7 to intron 11 of NLRP2 and approximately 17 missenses have also been seen as single heterozygous mutations or variants in patients with recurrent and sporadic moles. However, the pathological significance of these single mutations or variants is still the subject of debate, and more data are needed to reach a conclusion on their potential involvement in the causation or genetic susceptibility for moles [19]. Interestingly, males with NLRP7 homozygous mutations have been found to have no reproductive problems, suggesting that NLRP7 is not required for normal spermatogenesis [20]. NLRP7 transcripts have been identified in several human tissues, including endometrium, placenta, haematopoietic cells, all oocytes stages, and preimplantation embryos. NLRP7 transcripts decrease after fertilisation and during preimplantation development to reach their lowest level at day 3 of embryonic development, which corresponds to the blastocyst stage, and then increase sharply from day 3 to day 5, which coincides with the transcriptional activation of the embryonic genome [21].
KHDC3L (KH domain containing 3-like), which was identified in 2011, is a second recessive gene responsible for RHMs. KHDC3L maps to chromosome 6, and available data indicate that this gene is a minor gene for RHMs, accounting for 10–14% of patients who do not have mutations in NLRP7 [22]. To date, four mutations in KHDC3L have been reported in patients with two defective alleles. KHDC3L transcripts have been identified in several human tissues, including all oocytes stages, preimplantation embryos, and haematopoietic cells. KHDC3L codes for a small protein of 217 amino acids belonging to the KHDC1 (KH homology domain containing 1) protein family, members of which contain an atypical KH domain that does not bind RNA as opposed to proteins with canonical KH domain. In humans, this family includes KHDC3L, KHDC1, DPPA5 (developmental pluripotency associated 5), and OOEP (oocyte-expressed protein). Expression of KHDC3L is highest in oocytes at the germinal vesicle stage and then decreases during preimplantation development and becomes undetectable at the blastocyst stage, similar to the expression prolife of NLRP7 [21]. In addition, KHDC3L co-localises with NLRP7 to the microtubule organising center and the Golgi apparatus in lymphoblastoid cell lines, which suggests that the two genes may have similar or overlapping functions in oocyte and early embryonic development [21, 23].
Genomic imprinting is an epigenetic regulatory process which allows the expression of one of the two alleles of an imprinted gene in a sex-specific pattern. Within the 30,000–40,000 genes in the human genome, the majority display a biallelic pattern of expression, whereas a small percentage of genes are known to be imprinted. This epigenetic alteration is not associated with a change in the sequence of the DNA; however, it modifies the function of the imprinted gene [24]. Imprinted genes are those which are expressed differently depending upon whether they are carried by chromosome of maternal or paternal origin. Usually, when a gene is paternally expressed, it is said to be maternally imprinted and vice versa. Genomic imprinting is a dynamic process, established and maintained mainly by DNA methylation, but also by other mechanisms like acetylation and histone modifications. More specifically, the non-active/not expressed copy of a gene is usually methylated, whereas the active/expressed copy is un-methylated. The ratio between the maternal and paternal genomes is crucial for the development of both embryonic and extra-embryonic tissues, with an excess of paternally derived chromosomes leading to a complete (no maternal genome) or partial (lower amount of maternal chromosomes) mole [21]. Importantly, this is very helpful for the accurate classification of HMs. More specifically, analyses of p57 expression by immunohistochemistry can be used, as p57 is the gene product of the paternally imprinted, maternally expressed gene, CDKN1C. CHMs, which lack a maternal genetic contribution, do not express p57 in villous cytotrophoblastic and villous stromal cells, while PHMs, which contain a maternal chromosomal complement, express p57 in these cells [4].
Imprinted genes are “correctly marked” in a sex-specific manner in the germline (primordial germ cells, PGCs). DNA methylation (inherited from the previous generation) is erased during migration of PGCs into the genital ridge and is re-established during gametogenesis, in sperm and growing oocytes (paternal and maternal imprinting pattern is established, respectively, for the next generation). After fertilisation (in zygote) and until the blastocyst stage although demethylation and extensive reprogramming occurs, imprints are maintained, thus survive erasure. For instance, during this period of global methylation changes, the methylated parental allele is protected against demethylation, whereas the unmethylated allele is protected against the acquisition of DNA methylation. In the blastocyst stage, a wave of de novo methylation occurs; however, imprints not only remain again unaffected but are also maintained in somatic cells and extra-embryonic tissues throughout the lifetime of the organism [25].
Studies of molar tissues have shown that DNA methylation across a number of genetic loci known to be maternally imprinted, was generally, though not universally, altered, indicative of a defective maternal imprinting. This contrasts to normal methylation patterns at paternal specific imprinted loci. These observations suggest failure of the oocyte to establish maternal epigenetic variations at imprinted loci. This results in an apparent paternal genotype or paternal “epigenotype” on the maternal allele of several imprinted genes and it is thought to be the mechanism through which a phenotype usually associated with androgenesis occurs [3]. Interestingly, molar tissue of familial BiCHM patients, with either NLRP7 or KHDC3L mutations, demonstrated imprinting defects with silencing of maternal imprinted genes and expression of paternal imprinted genes [26]. Imprinting loss has been reported to lead to choriocarcinoma, as well [27]. Thus, genomic imprinting confers functional differences between the paternal and maternal derived genomes, producing a requirement for a balanced biparental genetic representation in normal development, whereas disruption of normal balanced genomic imprinting in gestation may result in abnormal trophoblastic proliferation leading to molar pregnancy.
RHMs is not an endless condition: reproductive options in favour of a normal pregnancy
For patients with recurrent HM, reproductive options are currently limited. In some pedigrees, pregnancies with normal outcomes have been reported in affected women with BiCHMs [19], but in the majority of families, as in our study, there are no favourable pregnancy outcomes [3]. Akoury et al. reported three patients with two NLRP7 defective alleles that had a total of four live births from donated ova, whereas patients with two defective alleles in NLRP7 may have live births from spontaneous conceptions from their own oocytes in 1% of their pregnancies [19]. It is therefore of great importance that, women who present with RHMs and a positive family history, to be recognised by the obstetrician and gynaecologist and to be referred for suitable further investigation and counselling (Table 2). If the origin of a recurrent mole is androgenetic, then intracytoplasmic sperm injection (ICSI) can ensure monospermic fertilisation and avoid dispermic fertilisation. Pre-implantation genetic diagnosis (PGD) accompanied with fluorescent in situ hybridisation (FISH) can select against the transfer of 46,XX embryos, preventing CHMs resulting from a fertilisation of an inactive oocyte, by a haploid X-bearing spermatozoon which subsequently duplicates.
Preimplantation confirmation of diploidy by FISH also avoids the selection of triploid PHMs that may result from mechanisms other than dispermic fertilisation [28]. In cases of diploid biparental moles that do not contain excess of paternally inherited genetic material, but have mutations in NLRP7 or KHDC3L genes that disrupt normal imprinting and have been associated with a defect in the epigenetic status of the oocyte genome, oocyte donation should be mainly considered in order to achieve normal pregnancy [19, 29,30,31]. This strategy was encouraged for our patient too, who is now considering oocyte donation as the most preferable management in order to achieve pregnancy. Recently, genome editing is progressing so that may become also a promising option for BiCHM to correct gene mutations in germinal vesicle stage oocytes. For example, clustered, regularly interspaced, short palindromic repeat/cas 9 (Cripsr/Cas9) has emerged as a highly efficient new tool to edit genomic sequences and to correct mutated genes. Such discoveries pave the way for improved targeted personalised medicine for rare conditions, such as BiCHM [32].
Conclusions
Cases of molar pregnancies although rare are of crucial importance to be early recognised and to be appropriately managed. The mechanisms leading to a molar pregnancy is a complex genetic game of chess. At a clinical level, patients with RHMs cannot be distinguished from non-recurrent sporadic moles. Thus, DNA testing should be performed. When HMs are of biparental origin, with familial clustering and NLRP7 or KHDC3L mutations are detected, the option of oocyte donation is currently the best reproductive strategy which can robustly support that this condition is not endless and due to reproductive advances in the field of IVF, the possibility of a normal pregnancy can be an achievable goal.
References
Seckl MJ, Sebire NJ, Berkowitz RS. Gestational trophoblastic disease. Lancet. 2010;376:717–29.
Lurain JR. Gestational trophoblastic disease I: epidemiology, pathology, clinical presentation and diagnosis of gestational trophoblastic disease, and management of hydatidiform mole. Am J Obstet Gynecol. 2010;203:531–9.
Williams D, Hodgetts V, Gupta J. Recurrent hydatidiform moles. Eur J Obstet Gynecol Reprod Biol. 2010;150:3–7.
Hui P, Buza N, Murphy KM, Ronnett BM. Hydatidiform moles: genetic basis and precision diagnosis. Annu Rev Pathol. 2017;12:449–85.
Tuncer ZS, Bernstein MR, Wang J, Goldstein DP, Berkowitz RS. Repetitive hydatidiform mole with different male partners. Gynecol Oncol. 1999;75:224–6.
Wolfberg AJ, Berkowitz RS, Goldstein DP, Feltmate C, Lieberman E. Postevacuation hCG levels and risk of gestational trophoblastic neoplasia in women with complete molar pregnancy. Obstet Gynecol. 2005;106:548–52.
Carey L, Nash BM, Wright DC. Molecular genetic studies of complete hydatidiform moles. Transl Pediatr. 2015;4:181–8.
Ambani LM, Vaidya RA, Rao CS, Daftary SD, Motashaw ND. Familial occurrence of trophoblastic disease—report of recurrent molar pregnancies in sisters in three families. Clin Genet. 1980;18:27–9.
La Vecchia C, Franceschi S, Fasoli M, Mangioni C. Gestational trophoblastic neoplasms in homozygous twins. Obstet Gynecol. 1982;60:250–2.
Parazzini F, La Vecchia C, Franceschi S, Mangili G. Familial trophoblastic disease: case report. Am J Obstet Gynecol. 1984;149:382–3.
Seoud M, Khalil A, Frangieh A, Zahed L, Azar G, Nuwayri-Salti N. Recurrent molar pregnancies in a family with extensive intermarriage: report of a family and review of the literature. Obstet Gynecol. 1995;86:692–5.
Moglabey YB, Kircheisen R, Seoud M, El Mogharbel N, Van den Veyver I, Slim R. Genetic mapping of a maternal locus responsible for familial hydatidiform moles. Hum Mol Genet. 1999;8:667–71.
Kircheisen R, Ried T. Hydatidiform moles. Hum Reprod. 1994;9:1783–4.
Sensi A, Gualandi F, Pittalis MC, Calabrese O, Falciano F, Maestri I, et al. Mole maker phenotype: possible narrowing of the candidate region. Eur J Hum Genet. 2000;8:641–4.
Fisher RA, Hodges MD, Newlands ES. Familial recurrent hydatidiform mole: a review. J Reprod Med. 2004;49:595–01.
Van den Veyver IB, Al-Hussaini TK. Biparental hydatidiform moles: a maternal effect mutation affecting imprinting in the offspring. Hum Reprod Update. 2006;12:233–42.
Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38:300–2.
Slim R, Wallace EP. NLRP7 and the genetics of hydatidiform moles: recent advances and new challenges. Front Immunol. 2013;4:242.
Akoury E, Gupta N, Bagga R, Brown S, Déry C, Kabra M, et al. Live births in women with recurrent hydatidiform mole and two NLRP7 mutations. Reprod BioMed Online. 2015;31:120–4.
Qian J, Deveault C, Bagga R, Xie X, Slim R. Women heterozygous for NALP7/NLRP7 mutations are at risk for reproductive wastage: report of two novel mutations. Hum Mutat. 2007;28:741.
Nguyen NM, Slim R. Genetics and epigenetics of recurrent hydatidiform moles: basic science and genetic counselling. Curr Obstet Gynecol Rep. 2014;3:55–64.
Parry DA, Logan CV, Hayward BE, Shires M, Landolsi H, Diggle C, et al. Mutations causing familial biparental hydatidiform mole implicate C6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am J Hum Genet. 2011;89:451–8.
Akoury E, Zhang L, Ao A, Slim R. NLRP7 and KHDC3L, the two maternal-effect proteins responsible for recurrent hydatidiform moles, co-localize to the oocyte cytoskeleton. Hum Reprod. 2015;30:159–69.
Devriendt K. Hydatidiform mole and triploidy: the role of genomic imprinting in placental development. Hum Reprod Update. 2005;11:137–42.
Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem. 2011;286:18347–53.
El-Maarri O, Seoud M, Coullin P, Herbiniaux U, Oldenburg J, Rouleau G, et al. Maternal alleles acquiring paternal methylation patterns in biparental complete hydatidiform moles. Hum Mol Genet. 2003;12:1405–13.
Hashimoto K, Azuma C, Koyama M, Ohashi K, Kamiura S, Nobunaga T, et al. Loss of imprinting in choriocarcinoma. Nat Genet. 1995;9:109–10.
Reubinoff BE, Lewin A, Verner I, Safran A, Schenker JG, Abeliovich D. Intracytoplasmic sperm injection combined with preimplantation genetic diagnosis for the prevention of recurrent gestational trophoblastic disease. Hum Reprod. 1997;12:805–8.
Fisher RA, Lavery SA, Carby A, Abu-Hayyeh S, Swingler R, Sebire NJ, et al. What a difference an egg makes. Lancet. 2011;378:1974.
Rai L, Shripad H, Guruvayare S, Prashant A, Sunil A. Recurrent familial hydatidiform mole—a rare clinical problem. J Turk Ger Gynecol Assoc. 2012;13:284–6.
Sills ES, Obregon-Tito AJ, Gao H, McWilliams TK, Gordon AT, Adams CA, et al. Pathogenic variant in NLRP7 (19q13.42) associated with recurrent gestational trophoblastic disease: data from early embryo development observed during in vitro fertilization. Clin Exp Reprod Med. 2017;44:40–6.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kalogiannidis, I., Kalinderi, K., Kalinderis, M. et al. Recurrent complete hydatidiform mole: where we are, is there a safe gestational horizon? Opinion and mini-review. J Assist Reprod Genet 35, 967–973 (2018). https://doi.org/10.1007/s10815-018-1202-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-018-1202-9