
Abstract
Purpose
PIWI-interacting RNA (piRNA) is a sub-group of small RNAs about 30 nucleotides length which specifically expressed in mammalian germ cells. Although piRNAs play pivotal roles in spermatogenesis regulation, little is known in the testicular tissues of infertile men. To explore whether piRNA profile could serve as a biomarker for male infertility diagnosis in a clinic, in this study, we systematically investigated the expression profile of piRNAs in testicular tissues from the patients with non-obstructive azoospermia (NOA) between successful and unsuccessful sperm retrieval before micro-dissection testicular sperm extraction (micro-TESE).
Methods
The differential expression levels of piRNAs were evaluated using small RNA-Seq method. Ontologic analyses were performed to determine the presence of enriched biological processes.
Results
A total of 18,324 Homo sapiens piRNAs were identified by small RNA-Seq from NOA patient testicular tissues; among them, 959 piRNAs were significantly altered between successful and unsuccessful sperm retrieval groups, of which 951 testicular piRNAs were significantly downregulated and 8 piRNAs were upregulated in NOA patients with unsuccessful sperm retrieval (USR) groups compared to those with successful sperm retrieval (SSR) groups, respectively. Unexpectedly, 553 testicular piRNAs were found completely absent in USR but showing abundant in SSR, which suggests that those piRNAs might serve as a biomarker for micro-TESE application. A total of 20 significantly differential piRNAs (hsa-piR-20830, hsa-piR-4731, hsa-piR-6254, hsa-piR-419, hsa-piR-7152, hsa-piR-7548, hsa-piR-14195, hsa-piR-5026, hsa-piR-11482, hsa-piR-17765, hsa-piR-17102, hsa-piR-4484, hsa-piR-17260, hsa-piR-17098, hsa-piR-20511, hsa-piR-5802, hsa-piR-19121, hsa-piR-2510, hsa-piR-4745, hsa-piR-11873) were selected to further validate the RNA-Seq data by quantitative real-time polymerase chain reaction. In addition, bioinformatic analyses revealed that those altered piRNAs were involved in many important biological pathways, including apoptosis, cell proliferation, and differentiation.
Conclusions
Our results demonstrate that testicular tissues from NOA patients with successful and unsuccessful spermatozoa retrieval exhibit differential piRNA profiles. This study provides a useful resource to further elucidate the regulatory role of piRNAs in spermatogenesis and provides a profound clue to identify useful biomarkers for predicting residual spermatogenic loci in NOA patients during assisted reproductive treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Male infertility has become a worldwide reproductive health issue, according to the World Health Organization (WHO) estimates that the infertile couples closer to 80 million and a male factor contribute in approximately 50% of the cases [1, 2]. In fact, the cause remains idiopathic in most of male infertility cases; thus, it is necessary to seek the potential biomarkers and mechanism for the idiopathic azoospermia diagnosis in a clinic. Non-obstructive azoospermia (NOA) which is caused by spermatogenesis failure accounts for about 60% of the azoospermia and 10% of male infertility [3]. Although the testicular function is poor and no any spermatozoa could be found in ejaculated semen from NOA patients, there may still be a focal area of spermatogenesis in some patients’ testes [4]. Patients with NOA can also have a healthy offspring as long as spermatozoa could be retrieved from their testes through testicular spermatozoa extraction surgery followed by combination with intracytoplasmic sperm injection (ICSI). However, how to search these focal spermatozoa more accurately remains an open question in recent years. In 1998, micro-dissection of testicular sperm extraction (micro-TESE) was applied to clinical testicular sperm retrieval because of its advantages of small injury, high pertinence, and high rate of sperm detection [5]. While the fact is not always the case, according to the reports in the literatures, micro-TESE take fine acceptable recovery rate was 35–77%, which can also cause great damages to the testis, especially for those people who cannot successfully retrieve sperm after the surgery [6]. Therefore, it is necessary for us to find out a non-invasive diagnostic technique which can predict the presence of sperm into the testis so that a large number of NOA patients would avoid great pains from the surgical interventions.
Spermatogenesis is a highly complex process of differentiation, including self-renewal and proliferation of spermatogonia, meiotic division of spermatocytes, and post-meiotic differentiation of spermatids into spermatozoa. These events are regulated by a variety of genetic and epigenetic factors, and the function of regulating non-coding small RNAs on spermatogenesis is gaining more and more attentions in the recent decade [7, 8]. Occupying 98.5% of non-protein-coding sequence in human genome, the vast majority are transcribed into regulatory non-coding RNAs, which are closely related to several human diseases such as infertility. Male germ cells express several classes of non-coding small RNAs, including Dicer-dependent miRNAs and endogenous small interfering RNAs (endo-siRNAs), as well as Dicer-independent PIWI-interacting RNAs (piRNAs) [9, 10]. miRNAs are a family of short (20–23 nucleotides), single-stranded small non-coding RNA molecules that are required for regulating post-transcriptional gene silencing through a base pair binding on their target mRNAs, thereby inducing translational inhibition or repression [11], and it is conceivable that any deregulation in miRNA expression patterns would significantly affect spermatogenesis pathways and lead to several types of reproductive abnormalities [12]. piRNAs are found as a new type of small non-coding RNAs (24–30 nucleotides) in Drosophila melanogaster, mice, rat, and human species male germ cells in 2006 [13,14,15,16]. Interestingly, piRNAs are divided into two classes of piRNAs: prepachytene and pachytene piRNAs, which are generated in the testes of mammals [17]. With the deepening of the research, a large number of literatures reporting piRNAs are involved in regulation of reproduction and play an important role in the process of spermatogenesis; one of the most important function is to control the activity of genome genetic components such as transposons and repetitive sequence, ensuring genomic stability and integrity of the germ line cells [18].
To date, several PIWI proteins are found to be involved in piRNA biogenesis pathway, such as MIWI (PIWIL1), MILI (PIWIL2), and MIWI2 (PIWIL4) in mice (human). All of them are predominantly expressed in the gonad, and MIWI is associated with pachytene piRNA biogenesis, whereas MIWI2 is responsible for prepachytene piRNA pathway and MILI is related to both prepachytene and pachytene piRNA cluster generation [17]. Given that PIWI proteins are germ line-specific protein, the loss of function of those PIWI proteins should be resulted in individual infertility in both mice and human. Indeed, Mili- or Miwi2-deficient male mice exhibit meiotic prophase I defects that are attributed to genetic damages caused by de-suppression of retrotransposon activity in the absence of a piRNA silencing mechanism, and Miwi KO male mice exhibited a spermiogenic arrest at step 4 [19,20,21]. More striking, Gou et al. recently reported that germ line mutations in human Piwi (Hiwi) can result in human azoospermia by impairing histone-to-protamine exchange during human spermiogenesis, which is the first time to validate the function of PIWI proteins in human fertility [22]. Therefore, a comprehensive analysis of testicular-specific piRNAs will help to understand the mechanisms by which these piRNAs coordinate their target genes to regulate spermatogenesis, thus helping to understand the causes for NOA.
Our goals, in this study, are to dissect the piRNA-differentiated expression profiles between successful sperm retrieval (SSR) and unsuccessful sperm retrieval (USR) groups and investigate whether the differential piRNAs in testicular tissues could be served as the potential biomarkers for NOA patient. Our data provides a useful resource to further elucidate the regulatory role of piRNAs in spermatogenesis and provides a profound clue to identify useful biomarkers for predicting residual spermatogenic loci in NOA patients.
Materials and methods
Testicular sample collection and processing
All samples were abandoned biopsy testicular tissue after treatment and collected according to protocols approved by the Medical Ethics Committee from the Center for Reproductive Medicine, Tongji Medical College, Huazhong University of Science and Technology. All patients signed informed consent for the collection and use of their samples for this study. Testicular samples were obtained from 10 patients (aged 22–30 years) with non-obstructive azoospermia who underwent micro-TESE at the Center for Reproductive Medicine, Huazhong University of Science and Technology. The samples were classified into two groups according to SSR (n = 5) or USR (n = 5) in the surgical procedures of micro-TESE. Immediately after retrieval, testicular tissues were snapped frozen and stored in liquid nitrogen at − 196 °C until processed. Briefly, we usually collected three sets of biopsies when micro-TESE was performed in each NOA patient who wanted to be treated by ICSI. Among these biopsies, one was used for clinical detection under microscopy to search mature spermatozoa for ICSI, another one was snapped frozen into liquid nitrogen for RNA-Seq analyses, and the last one was fixed in Bouin’s solution for histological evaluation. If we found the live sperm into the first biopsy, we classified this patient into SSR groups; otherwise, he will be recruited into USR groups. The seminiferous tubular histological analysis results of all biopsy testicular samples from 10 NOA patients (both SSR and USR groups) were presented in Fig. 1. The detailed clinical information of the patients is showed in Table 1.

Histological analyses of testis biopsy tissues from 10 recruited NOA patients in this study. Upper panel showing the PAS-staining results of 5 NOA patients in USR groups. Lower panel showing the PAS-staining results of 5 NOA patients in SSR groups. Scale bar = 100 μm
Written informed consent was obtained from all patients. The study was approved by the ethics committees of Tongji Medical College, Huazhong University of Science and Technology.
Isolation of total RNA and small RNA-Seq
Total RNAs were extracted from the human testis derived from SSR and USR groups using TRIZOL (Invitrogen) following the manufacturer’s procedure. RNA quality was verified with the use of Agilent 2100 Bioanalyzer Eukaryote Total RNA Pico assay (Agilent Technologies). High-quality total RNA should be used as starting material and the RNA integrity number (RIN) > 7 is recommended. After polyacrylamide gel electrophoresis (PAGE) purification of RNA molecules smaller than 45 nucleotides, the 5′ and 3′ RNA adaptors were ligated to the 5′ and 3′ ends of the RNAs. The small RNA molecules were then transcribed to single-stranded cDNA and amplified using the adaptor primers. The PCR products were supplied for the cluster generation and sequenced using Illumina HiSeq 2500 sequencing systems (Illumina, San Diego, CA, USA). To identify piRNAs in human testis tissues, genome-wide comparison of the sequences was conducted with the testis referenced genome sequences and all of the testis-specific piRNAs were existed in the piRNA bank (http://pirnabank.ibab.ac.in/). The DESeq software used negative binomial distribution and a shrinkage estimator for the distribution’s variance to detect differential expression of small RNAs from high-throughput sequencing assay [23].
Bioinformatic analysis for piRNAs
The raw data is subjected to a high throughput of the sample via the single-ended mode of the primer sequencing platform. After removing the primer, the adapter, and the test sequence, the quality of the base is measured and the length is screened for the final reliable sequence. After that, the occurrence of each unique read was counted as tags. These unique tags were mapped to the human genome (GRCh37.p5) using SOAP2.0 [24], and then the tags within one mismatch were selected for further analysis while not matched tags in the piRNA bank against the sequences of small non-coding RNAs (rRNA, tRNA, snRNA, snoRNA) available on Rfam [25] were submitted to the subsequent matching steps. Meanwhile, known piRNAs in Homo sapiens are identified in the piRNA bank (http://pirnabank.ibab.ac.in/) [26]. GenBank non-coding RNA database (http://biobases.ibch.poznan.pl/ncRNA/), repeats database, coding region of reference genome [27] and to classify these tags into other small non-coding RNA, mRNA, genomic repeats, or unclassified tags if they were not assigned to any of the above databases. Meanwhile, the length distribution of small RNA was analyzed. In general, the length of the small RNA range of 18~30 nt and the peak energy of the length distribution helped us to determine the type of small RNA, such as miRNA concentrated in 21 or 22 nt, siRNA concentrated in 24 nt, and piRNA concentrated in 30 nt [27]. The global statistics and quality controls are presented in Supplemental Fig. S1a-c.
Analysis of significant differential piRNAs
For each sample, the sequence was compared to the new predicted piRNA library. The piRNA expression was calculated using the TPM calculation transcript per million, TPM formula = (the number of readings per piRNA) / (sample ratio on the read number) × 106; TPM meaning is the proportion of each pair of paired pairs of piRNA expression indicators, which is better than the number of pairs of readings for the normalized expression value. Both normalization and differential expression analyses were performed using DESeq2 (significant changes were defined as p value < 0.05) [28]. Log2 values of fold change (twofold) of dysregulated piRNAs in USR groups were plotted against their counts in SRS groups using the R script of the ggplot2 package.
Quantitative RT-qPCR analysis
Quantitative reverse-transcription polymerase chain reaction (RT-qPCR) was performed to validate the small RNA-Seq data. Eleven well-known piRNAs were selected to be analyzed by RT-qPCR. Reverse transcriptase reactions contained 100 ng of purified total RNA, 5 μl 5 × PAP/RT buffer, 1 μl 2.5 U/μl Poly A Polymerase, and 1 μl RTase Mix and RNase/DNase-free ddH2O. Each reaction mixture contained 10 μl 2X All-in-One PCR mix, 2 μl 2 μM All-in-One™ miRNA qPCR Primer, 2 μl 2 μM Universal Adapter PCR Primer, 2 μl cDNA, and deionized water to a total volume of 20 μl. Reactions were run with the following thermal cycling parameters: 95 °C for 10 min (predenaturation), followed by 40 cycles of 95 °C for 10 s (denaturation), 60 °C for 20 s (annealing), and 72 °C for 10 s (extending). The threshold cycle (Ct) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold, and each sample was normalized on the basis of its endogenous RNA U6b content. All primers were shown in Table S1.
Statistical analysis
The statistical analysis was performed using SPSS 21.0 software. Group data are expressed as means ± standard deviation (SD). The DESeq statistics were used to identify differentially expressed piRNAs between SSR and USR groups. The differences between the two groups were analyzed using Student’s t test. A result of p value < 0.05 from a two-tailed test was interpreted to indicate a significant difference. For validating the RNA-Seq results, we performed RT-qPCR and the relative quantitative method of 2−ΔCq to measure the dynamic change of specific selected piRNA.
Results
Small RNA-Seq reveals differential piRNA profiles in NOA patients
To compare the piRNA profiles of testicular tissues from NOA patients with successful and unsuccessful micro-TESE sperm retrieval, we performed small RNA deep sequencing to identify piRNA profiles and compared the piRNA expression profile in those two groups (SSR and USR group, respectively). A total of 18,324 Homo sapiens piRNAs were identified by small RNA-Seq from NOA patient testicular tissues (Table S2) and the piRNA expression abundance was showed in Fig. S1d. Among them, we found that 959 piRNAs were significantly changed (fold change > 2, p value < 0.05) between USR and SSR groups, and 951 piRNA expression levels were downregulated, whereas only 8 piRNAs were upregulated in USR groups compared to those in SSR groups (Fig. 2a, b, Table S3). Of which, 553 piRNAs were completely absent in USR groups and exclusively expressed in NOA patients with SSR groups (Fig. 2c), suggesting those piRNAs may serve as indicative biomarkers to predict sperm retrieval successfully before micro-TESE. Interestingly, we found that the piRNA expression levels displayed higher homogeneity among 5 NOA patients in the USR group, whereas these exhibited a great variation among the NOA patients in the SSR group, which are consistent with the testicular tubule histology between USR and SSR group patients (Fig. 1). This is caused, at least in part, by the inhomogeneity of residual spermatogenic loci among SSR group patients.

RNA-Seq reveals a differential piRNA expression profiles. a Heat map showing 959 piRNA expression levels that are significantly changed in NOA patients with unsuccessful sperm retrieval (USR) groups compared to those of successful sperm retrieval (SSR) groups. The Arabic number indicates different RNA-Seq data from an individual patient. b Volcano plots showing the differential expression levels of the total identified piRNAs. FDR means false discovery rate, T/C means test/control, test represents USR groups, and control represents SSR groups. c Venn diagram showing the significantly differentially expressed piRNAs shared or unique numbers among USR and SSR groups
Validation of RNA-Seq results by RT-qPCR
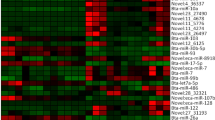
To validate the RNA-Seq data, we performed RT-qPCR to verify 20 significantly changed (top 30 changed) piRNA (hsa-piR-20830, hsa-piR-4731, hsa-piR-6254, hsa-piR-419, hsa-piR-7152, hsa-piR-7548, hsa-piR-14195, hsa-piR-5026, hsa-piR-11482, hsa-piR-17765, hsa-piR-17102, hsa-piR-4484, hsa-piR-17260, hsa-piR-17098, hsa-piR-20511, hsa-piR-5802, hsa-piR-19121, hsa-piR-2510, hsa-piR-4745, hsa-piR-11873) expression levels between USR and SSR groups. All qRT-PCR experiments were performed in triplicate and the results (Cq values) were normalized to RNA U6b. The RT-qPCR fold change results showed that all of the selected piRNA expression levels are significantly decreased in USR testicular tissues relative to SSR ones (p value < 0.05), which are largely consistent with the RNA-Seq data (Fig. 3a and Table S3). To determine whether the testicular piRNAs were expressed in the seminal plasma as well, we detected those 20 significantly differential piRNA expression levels in the seminal plasma by RT-qPCR and found that two piRNA (hsa-piR-6254 and has-piR-17765) expression levels in the seminal plasma are comparable with those of testicular tissues (Fig. 3b), suggesting these two piRNAs that exist in seminal plasma may serve as non-invasive biomarkers for predicting testicular spermatozoa retrieval before micro-TESE.

qRT-PCR analyses of 20 significantly changed piRNAs to validate RNA-Seq. a Comparison of the 20 most significantly changed piRNA expression levels among obstructive azoospermia (OA) groups, USR groups, and SSR groups by qRT-PCR. b qRT-PCR analyses of 20 significantly changed piRNA expression levels in human seminal plasma and testicular tissues. Data are shown as mean ± SEM (n = 5)
GO and KEGG analyses
To discover the underline functions of piRNAs during spermatogenesis, we tried to further perform GO and KEGG analyses using our small RNA-Seq data. Tot score ≥ 10 and Tot energy ≤ − 30 target genes were selected for the analyses. According to the p value less than or equal to 0.05 that was defined as a significant difference in GO term (Table S4), we found that more than 10 GOs were significantly higher in each GO category, and the top three GO categories are shown as the biological process, molecular function, and cellular component (Fig. 4). Apart from GO analysis, KEGG was used to construct a pathway enrichment of predicted piRNA target genes. Total 31 KEGG pathways were identified based on our RNA-Seq data, and many signaling pathways, such as PI3K-Akt signaling pathway and MAPK signaling pathway, were reported to closely associate with spermatogenesis (Fig. 5 and Table S5). Additionally, the top 12 biological processes, including development, cell motility, and cell communication, were linked with piRNAs followed by KEGG analyses, indicating those testicular piRNAs play multiple functions during spermatogenesis.

Gene ontology term analyses reveal that those significantly altered piRNAs are mainly involved in three GO categories, including the biological process, molecular function, and cellular component

Diagram showing the significant enrichment KEGG pathway. A total of 20 pathways exhibited a significant alteration
Discussion
At present, there are no reliable molecular makers to predict the chances of successful sperm retrieval in NOA patients. Alteration of piRNA expression profiles in testicular tissues between successful and unsuccessful sperm retrieval NOA patients can represent the residual spermatogenesis capacity to some extent and may have the potential to be used as new biomarkers for prediction before micro-TESE. In this study, for the first time, we applied the high-throughput technology Illumina Hiseq platform to compare the testicular piRNA profiles between USR and SSR groups, and validate a set of piRNAs by individual qRT-PCR towards the development of clinically practicable biomarkers for predicting the outcomes of micro-TESE. As a result, 553 piRNAs are exclusively expressed in NOA patients with spermatozoa that were successfully retrieved, indicating those piRNAs may have functions for regulation of spermatogenesis in human and also be considered as the potential biomarkers to predict sperm retrieval outcomes. In fact, the alteration of piRNA profiles in seminal plasma has been reported that those altered piRNAs can be predicted azoospermia and asthenozoospermia symptom in infertile men [29]; however, the remaining questions about piRNA expression profiles in NOA patient testicular tissues have not yet been addressed so far. Hence, our study has been directly reflected the local spermatogenic state in different NOA patients and further deciphered the piRNA differential expression profiles in NOA patients with successful or unsuccessful testicular spermatozoa retrieval.
In recent years, RNA research has become an important hotspot in the field of life science, as a new type of small RNA, piRNA, has received widespread attention. Currently, the molecular basis of male NOA symptom remains largely unknown, and the role of piRNAs in human spermatogenesis has not been well documented. A recent study has demonstrated that the piRNA machinery has a siRNA-like function in mouse testes and plays a central role in male germ cell development and maturation [30]. Moreover, piRNAs have an antisense orientation to transposon transcripts and induce silencing by hybridizing with them. Due to the diverse and pivotal functions of piRNAs in the male reproductive system, dysregulation and dysfunction of piRNA profiles usually lead to male infertility. Therefore, we speculate that the normal piRNA profile in testicular tissues indicating with a well spermatogenesis and the specific testicular piRNA may serve as biomarkers for predicting NOA patient sperm retrieval outcome through micro-TESE surgery. However, in our study, it is notable that the piRNA complement of the two groups differs due to the presence of partial germ cells in SSR groups but the absence in USR groups. Given this, a great variation of piRNA expression was observed from different NOA patients in SSR groups and the testis histological analyses further corroborated that as well (Fig. 1).
Importantly, in this study, we found that the targets for the most abundant piRNAs were predicted using NCBI blast package and the detailed information was summarized in Supplemental Fig.S2-5. As shown in Supplemental Fig.S5, most of the potential targets appeared to be retrotransposons, which were consistent with previous report [18]. Although piRNA pathways have been supposed to be repressed transposons in genome, and control meiosis process, cell apoptosis, and male fertility, there are many questions waiting for us to solve. For instance, what is the starting factor of the piRNA cluster that transcribes the piRNA precursor? Whether piRNA’s regulation of protein-coding genes is in the development of the reproductive system or is a time period? And how does the stimulus of the external environment affect the production of piRNA? Through the exploration of these questions, we will develop a deeper and accurate understanding of the reproductive issues in human. Our findings may provide additional direction to address the above questions regarding the molecular mechanism of NOA symptom in humans, which contribute to the field of male infertility.
In summary, we identified altered piRNA profiles between the NOA patient testicular tissues with and without successfully retrieved spermatozoa by micro-TESE. Several piRNA clusters were examined to better distinguish NOA patients who cannot find sperm after micro-TESE. However, further studies and more recruits in similar populations of NOA patient testicular tissues are needed to confirm some of the scenarios presented in this study. In current context, it would also be of great interest to screen the genetic expression in human testicular tissues of the same NOA patients because it would contribute to the elucidation of which kind of gene expression regulation is exerted by those piRNAs and at what level. Additionally, once we test the selected piRNAs in the seminal plasma with significant difference between USR and SSR groups in the future, the differential ones could predict to retrieve sperm with mere slightly hope. To some extent, our findings provide novel differential piRNAs which could be served as valuable biomarkers to predict the focal spermatogenic loci for micro-TESE surgery in NOA patients.
References
Inhorn MC, Patrizio P. Infertility around the globe: new thinking on gender, reproductive technologies and global movements in the 21st century. Hum Reprod Update. 2015;21:411–26.
Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. 2011;25:271–85.
Donoso P, Tournaye H, Devroey P. Which is the best sperm retrieval technique for non-obstructive azoospermia? A systematic review. Hum Reprod Update. 2007;13:539–49.
Turek PJ, Ljung BM, Cha I, Conaghan J. Diagnostic findings from testis fine needle aspiration mapping in obstructed and nonobstructed azoospermic men. J Urol. 2000;163:1709–16.
Schlegel PN, Li PS. Microdissection TESE: sperm retrieval in non-obstructive azoospermia. Hum Reprod Update. 1998;4:439.
Colpi GM, Piediferro G, Nerva F, Giacchetta D, Colpi EM, Piatti E. Sperm retrieval for intra-cytoplasmic sperm injection in non-obstructive azoospermia. Minerva Urol Nefrol. 2005;57:99–107.
Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H. Stem cell division is regulated by the microRNA pathway. Nature. 2005;435:974–8.
Maatouk DM, Loveland KL, McManus MT, Moore K, Harfe BD. Dicer1 is required for differentiation of the mouse male germline. Biol Reprod. 2008;79:696–703.
Yadav RP, Kotaja N. Small RNAs in spermatogenesis. Mol Cell Endocrinol. 2014;382:498–508.
Kotaja N. MicroRNAs and spermatogenesis. Fertil Steril. 2014;101:1552–62.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Papaioannou MD, Nef S. microRNAs in the testis: building up male fertility. J Androl. 2010;31:26–33.
Grivna ST, Beyret E, Wang Z, Lin HF. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–14.
Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202.
Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, et al. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–7.
Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–7.
Fu Q, Wang PJ. Mammalian piRNAs: biogenesis, function, and mysteries. Spermatogenesis. 2014;4:e27889.
Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, et al. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 2006;20:1732–43.
Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, et al. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell. 2007;12:503–14.
Reuter M, Chuma S, Tanaka T, Franz T, Stark A, Pillai RS. Loss of the Mili-interacting Tudor domain-containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat Struct Mol Biol. 2009;16:639–46.
Deng W, Lin H. miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev Cell. 2002;2:819–30.
Gou LT, Kang JY, Dai P, Wang X, Li F, Zhao S, et al. Ubiquitination-deficient mutations in human piwi cause male infertility by impairing histone-to-protamine exchange during spermiogenesis. Cell. 2017;169:1090.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11
Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–7.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–8.
Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–4.
Tyner C, Barber GP, Casper J, Clawson H, Diekhans M, Eisenhart C, et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017;45:D626–34.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15
Hong Y, Wang C, Fu Z, Liang H, Zhang S, Lu M, et al. Systematic characterization of seminal plasma piRNAs as molecular biomarkers for male infertility. Sci Rep. 2016;6:24229.
Zhang P, Kang JY, Gou LT, Wang J, Xue Y, Skogerboe G, et al. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res. 2015;25:193–207.
Funding
This work was supported, in part, by grants from the National Natural Science Foundation of China (No. 31671551) and Independent Innovation Cross Team Foundation of Huazhong University Science and Technology (No. 2016JCTD119).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Written informed consent was obtained from all patients. The study was approved by the ethics committees of Tongji Medical College, Huazhong University of Science and Technology.
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Cao, C., Wen, Y., Wang, X. et al. Testicular piRNA profile comparison between successful and unsuccessful micro-TESE retrieval in NOA patients. J Assist Reprod Genet 35, 801–808 (2018). https://doi.org/10.1007/s10815-018-1134-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-018-1134-4