
Abstract
Purpose
To determine the molecular basis of familial, autosomal-recessive, non-obstructive azoospermia in a consanguineous Iranian Jewish family.
Methods
We investigated the genetic cause of non-obstructive azoospermia in two affected siblings from a consanguineous family. Homozygosity mapping in the DNA samples of the patients and their normospermic brother was followed by exome analysis of one of the patients. Other family members were genotyped for the mutation by Sanger sequencing. The mutation effect was demonstrated by immunostaining of the patients’ testicular tissue.
Results
The two patients were homozygous for a splice site mutation in SYCE1 which resulted in retention of intron three in the cDNA and premature stop codon. SYCE1 encodes a Synaptonemal Complex protein which plays an essential role during meiosis. Immunostaining of patient’s testicular tissue with anti-Syce1 antibody revealed an undetectable level of Syce1. Histological examination of the patients’ tissue disclosed immature-stages spermatocytes without mature forms, indicating maturation arrest.
Conclusion
The significance of most synaptonemal complex proteins was previously demonstrated in a mutant mouse model. The present report underscores the importance of synaptonemal complex proteins in spermatogenenesis in humans. Our new approach, combining homozygosity mapping and exome sequencing, resulted in one of the first reports of an autosomal-recessive form of NOA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Male infertility accounts for 30–55 % of infertile couples. This is manifested by the high rate of men of reproductive age, 1 in 13, who require medical assistance in order to have a child [1]. Azoospermia, which is the complete absence of sperm in the ejaculate, accounts for 10–15 % of male infertility cases and generally affects 1 % of the male population [2]. Azoospermia is classified as obstructive or non obstructive (NOA). Obstructive azoospermia can result from blockage anywhere in the ductal system as a consequence of severe infection, iatrogenic injury or congenital anomalies.
Non obstructive azoospremia accounts for 60 % of cases of azoospermia and is caused by intrinsic testicular disease (primary testicular failure) or secondary testicular failure (endocrinopathy or other condition suppressing sperm production).
The evaluation for azoospermia includes endocrine tests and urologic and genetic evaluation. Currently the three most relevant genetic causes of azoospermia are: mutation in the CFTR gene (associated with congenital bilateral absence of the vas deferens and obstructive azoospermia), chromosomal abnormalities (such as Klinefelter syndrome) and microdeletions on chromosome Y, causing NOA [3]. Even after complete evaluation most cases of azoospermia are defined as idiopathic. Unfortunately, no medical treatment has proven reliably effective for improving semen parameters or fertility in men with idiopathic subfertility.
It has been previously proposed that azoospermia may result from single-gene disorders including haploinsufficiency of the Synaptonemal Complex protein SYCP3 [4]. Ayhan et al. recently published their findings regarding two mutations causing azoospermia in two consanguineous families [5]. It is reasonable to assume that additional genetic abnormalities might also explain male infertility.
Synaptonemal Complex Central Element 1 (Syce1) is a major component of the synaptonemal complex (SC), which is formed between homologous chromosomes during meiotic prophase and exists only during the first meiotic division [6]. Male Syce1 null mice are infertile, have abundant primary spermatocytes, and lack postmeiotic cells [7]. In women, haploinsufficiency of SYCE1 has been implicated in cessation of menstrual cycles in premature ovarian failure [8] while nonsense homozygous mutation in SYCE1 was shown to cause premature ovarian failure in two daughters of a consanguineous family [9].
We report the results of molecular investigation of two azoospermic males originating from a consanguineous family linking human mutation in SYCE1 with NOA. The results are followed by immunostaining of testicular tissue to support our findings.
Materials and methods
Patients
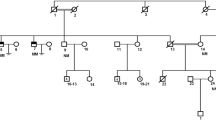
The subjects of the study, individuals II-2 and II-3, originated from a consanguineous (first cousins) Iranian-Jewish couple (Fig. 1). Both were referred for evaluation because of failure to reproduce. Their physical examination was unremarkable: height, weight and hair distribution were normal; testes were of normal size, volume and texture and no varicocelle was noted. Hormone profile including FSH, LH, and testosterone was also within normal limits. Sperm testing disclosed complete azoospermia. Further evaluation showed normal karyotype and analysis for deletions in the Y chromosome was negative. In both patients, testicular fine needle aspiration (TEFNA), repeated 20 times for each testis, revealed only primary spermatocytes without mature forms, indicating azoospermia due to maturation arrest (Fig. 2). In view of the complete absence of viable mature sperm both couples required sperm donation.

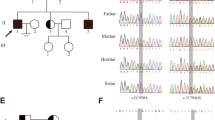
Family pedigree and the effect of the SYCE1 mutation. The patients are represented by filled symbols and the genotype at the mutation site is shown. The DNA sequence flanking the c.197-2 A > G mutation (arrow) in the SYCE1 gene is shown in the DNA samples of a patient (b), a carrier (c), and a healthy control (d)

H&E of patient II-3. H&E staining on paraffin sections of TEFNA taken from patient II-3 showing complete meiotic maturation arrest. On the right, high magnification showing Sertoli cells (black arrowheads) and arrested spermatogonia (red arrowheads). As a result of the TEFNA procedure, the specimens were overall poor of cells; therefore the sections were assessed by two different pathologists to conclude the diagnosis
The family includes another healthy brother (II-4) and sister (II-1). The pedigree is shown in Fig. 1. Interestingly, the mother and sister in the family gave birth to their first offspring at the ages of 19 and 22, respectively, and the mother entered menopause at the age of 53.
All family members provided informed consent to participate in the study. The study protocol was approved by the Hadassah Medical Center and Ministry of Health Ethical Committees.
Homozygosity mapping
A search for common homozygous regions in the DNA of the affected siblings and their unaffected brother (II-4) was performed using the Affymetrix GeneChip Human Mapping 250 K SNP Array, as previously described [10]. In essence, this chip allows genotyping of SNPs with an average distance of ∼ 50 kb between the markers. Homozygous regions >5.0 cM were manually detected. We therefore searched for common homozygous regions in the patients’ DNA that were not shared by their fertile brother.
Whole exome sequencing
Whole exome sequencing, a technique for sequencing all the protein-coding genes in a genome (known as the exome), was performed on DNA from patient II-2 using the SureSelect Human All Exon V.2 Kit (Agilent Technologies, Santa Clara, California, USA) on HiSeq2000 (Illumina, San Diego, California, USA) as 100 bp paired-end runs. Reads alignment and variant calling were performed with DNAnexus software (Palo Alto, CA) using the default parameters with the human genome assembly hg19 (GRCh37) as a reference.
Immunohistochemistry
TEFNA of three controls and patients II-2 and II-3 were embedded in paraffin and 2 μm sections were immunostained using the BenchMark XT automated staining instrument (Ventana). The antibody used was anti SYCE1 (NBP1-88971, Novus) diluted 1:200.
Results
The two infertile brothers, II-2 and II-3, were affected with NOA; histopathological examination disclosed maturation arrest (Fig. 2). In light of parental consanguinity, we assumed a founder mutation transmitted in an autosomal-recessive manner. We therefore searched for common homozygous regions in the patients’ DNA, which were not shared by their unaffected brother, II-4. This analysis, performed with DNA SNP array, resulted in the identification of four regions larger than 2 Mb, chr3:133172767-147368416, chr7:7344050-9178462, chr10:127938705-135422505 and chrX:2690310-33162851 (numbering according to HG19). The regions, spanning a total of 53.9 Mb, encompassed hundreds of protein-coding genes. We therefore opted for whole exome sequencing in the DNA sample of patient II-2. Average exome coverage was x86.2 and 92.5 % of the exons were covered to > x7. The resultant variants were filtered and heterozygous, synonymous, low-covered (<x7), off target, and those present in dbSNP132 or the in-house dbSNP were removed. Thirty-seven variants survived this process, however only three of them resided within the shared homozygous regions and only one, chr10:135372457, c.197-2 A > G in the SYCE1 gene, was predicted pathogenic by Mutation Taster software [11]. The mutation was examined by Sanger sequencing in all family members and was found to segregate with the disease (Fig. 1). The mutation was absent from 55 Iranian Jewish fertile males and was not present in the 6503 exome analyses of healthy individuals available through the Exome Variant Server [NHLBI Exome Sequencing Project, Seattle, Washington, USA (http://evs.gs.washington.edu/ EVS-v.0.0.21) (Aug 30, 2013)].
The SYCE1 gene consists of 13 exons encoding 351 amino acids protein (GI:118573896). The c.197-2 A > G mutation disrupts the acceptor site of intron 3. Our findings could be confirmed either by generating cDNA from the patients’ testes, or employing immunohistochemistry at the protein level. Since the biopsy samples were embedded in paraffin in 2006, during clinical the evaluation of the patients, we were unable to generate cDNA from the testes. cDNA was generated from peripheral leukocytes (data not show) but was inconclusive. Immunostaining of testicular tissue from our patients using anti-Syce1 antibody disclosed undetectable levels of the Syce1 protein (Fig. 3). For validation purposes Western blot analysis on testis total protein extract yielded a single band at the predicted size of SYCE1 - ~50 kDa (Fig. 4).

Immunohistochemistry staining with anti-Syce1 antibody showing cross paraffin sections through the testicular seminiferous tubules of control (a) and patient II-2 (b). While in control sample Syce1 is localized to the chromatin at various spermatocytes maturation stages (inset in A highlighting a nucleus during pachytene stage), in the patient sample it is completely absent (arrested spermatogonia are marked by red arrowhead in B). Black arrowheads indicate Sertoli cells both in control and patient samples. Yellow arrowheads indicate late and mature spermatids, which do not express Syce1

Western blot. Normal testis total protein was extracted from TEFNA biopsy using radioimmunoprecipitation assay (RIPA) buffer. 40ug of protein was separated and analyzed by Western blot on 10 % SDS-PAGE and probed with anti-Syce1 Ab (1:1000; NBP1-88971, Novus). A single band at the predicted size of Syce1, ~ 50 kDa, is evident, underscoring the specificity of this antibody to Syce1 on testicular tissue
Discussion
Here we show for the first time that the human SYCE1 gene plays an essential role during meiosis. Syce1 is a major component of the synaptonemal complex (SC), which is formed between homologous chromosomes during meiotic prophase and exists only during the first meiotic division [6].
The significance of most SC proteins was previously demonstrated in mutant mouse models. These mice were invariably infertile, with the exception of female mutants in Sycp2 and Sycp3, which had litters of reduced size and increased aneuploidy [12]. The importance of SYCE1 for the more advanced stages of meiosis was underscored by the presence of early forms of spermatocytes with absence of mature forms in the testicular tissue of our patients (Fig. 3), consistent with maturation arrest. The male Syce1 null mice had abundant primary spermatocytes and lacked postmeiotic cells; arrested cells were likely eliminated by accelerated apoptosis [7]. However, in mice, and likely in humans, SYCE1 is essential for stable homologue interactions mediated by the SC and crossover formation.
Notably, apart from NOA, our patients, just like the Syce1 null mutant mice, were healthy and had no systemic symptoms, indicating that SYCE1 is required for meiosis only.
Haploinsufficiency of SYCE1 was implicated in very early cessation of menstrual cycles in one case of premature ovarian failure [8]. While nonsense homozygous mutation in SYCE1 was shown by Varies et al. to cause premature ovarian failure in two daughters of a consanguineous family [9], the normal menstrual cycles observed in the mother and sister of our patients, both heterozygous for the SYCE1 null allele, do not support haploinsufficiency of SYCE1 as a cause of premature ovarian failure.
The present report underscores the importance of SYCE1 for SC formation, completion of meiosis and fertility in humans. In order to estimate the contribution of SYCE1 mutations to human NOA, we determined the DNA sequence of the SYCE1 coding region in ten unrelated NOA patients of various ethnic origins. This analysis did not detect additional mutations in the gene, suggesting that SYCE1 mutations are not a common cause of NOA.
The main limitation of our study is our inability to support our finding by cDNA from the testes.
Our study is one of the first to present genetic causes linked to the mechanism of NOA in humans. Moreover it highlights the power of exome sequencing as a primary diagnostic tool that could be used to elucidate the genetic etiology of NOA in general.
References
Anderson JE, Farr SL, Jamieson DJ, Warner L, Macaluso M. Infertility services reported by men in the United States: national survey data. Fertil Steril. 2009;91:2466–70.
Jarvi K et al. CUA guideline: the workup of azoospermic males. Can Urol Assoc J. 2010;4:163–7.
Vogt PH, Edelmann A, Kirsch S, et al. Human Y chromosome azoospermia factors (AZF) mapped to different suregions in Yq11. Hum Mol Genet. 1996;5:933.
Miyamoto T et al. Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet. 2003;362:1714–9.
Ayhan O, Balkan M, Guven A, Hazan R, Atar M, Tok A, et al. Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia. J Med Genet. 2014. doi:10.1136/jmedgenet-2013-102102.
Page SL, Hawley RS. The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol. 2004;20:525–58.
Bolcun-Filas E et al. Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 2009;5:e1000393.
McGuire MM, Bowden W, Engel NJ, Ahn HW, Kovanci E, Rajkovic A. Genomic analysis using high-resolution single-nucleotide polymorphism arrays reveals novel microdeletions associated with premature ovarian failure. Fertil Steril. 2011;95:1595–600.
Vries LD, Behar DM, Smirin-Yosef P, Lagovsky I, Tzur S, Basel-Vanagaite L. Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J Clin Endocrinol Metab. 2014;25:jc20141268.
Edvardson S et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–62.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. Mutation Taster evaluates disease causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Schramm S et al. A novel mouse synaptonemal complex protein is essential for loading of central element proteins, recombination, and fertility. PLoS Genet. 2011;7:e1002088.
Acknowledgment
We wish to thank Vivi Zuri Naama Lexner and Olga Genin for excellent technical assistance.
Conflict of interest
The authors have no conflict of interest to declare.
Contributor statement
YC, BY, AS, HG, SZ, CR, OE and AF conceived and designed the experiments; EMS, YC, BY, AS, HG, and SZ performed the experiments; all the coauthors analyzed the data and participated in the writing of the paper; EMS undertook patient management, collection of samples, and delineation of the phenotype. The research was supported in part by a grant from the Joint Research Fund of the Hebrew University and Hadassah.
Author information
Authors and Affiliations
Corresponding author
Additional information
Capsule Splice site mutation in the SynaptonemaL Complex central Element 1 resulted in non obstructive azoopsermia in two siblings. The results confirmed by immunohystochemistry on testicular samples.
Rights and permissions
About this article

Cite this article
Maor-Sagie, E., Cinnamon, Y., Yaacov, B. et al. Deleterious mutation in SYCE1 is associated with non-obstructive azoospermia. J Assist Reprod Genet 32, 887–891 (2015). https://doi.org/10.1007/s10815-015-0445-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-015-0445-y