
Abstract
Purpose
This study evaluated and compared survival, re-expansion, and percentage of live cells of individual Days 5 and 6 human blastocysts that were vitrified and warmed with the Vit Kit Freeze/Thaw (Irvine Scientific, CA), or with two protocols using the Global Fast Freeze/Thaw Kits (LifeGlobal, Canada).
Methods
Frozen/thawed Day 2–3 or discarded embryos were cultured to blastocyst (culture day 5–6). Group 1 blastocysts were vitrified with the Vit Kit (n = 29) and High Security Vitrification (HSV) devices. Group 2 (n = 47) and Group 3 (n = 48) blastocysts were cryopreserved with the Global Fast Freeze Kit and 0.25 ml straws, using a direct plunge or a −100 °C holding step, respectively. Group 4 (Controls, n = 30) were not vitrified. Blastocysts were subsequently cultured for 24 h, assessed for survival and expansion, and then stained individually with propidium iodide and Hoechst. Live and total cell number was assessed with ImageJ (NIH), and the percentage of live cells calculated for each blastocyst.
Results
The percentage of live cells was not different between vitrified and control (non-vitrified) blastocysts, thus vitrification did not affect cell survival. Survival (following thawing and after 24 h culture), re-expansion, and percentage of live cells were not different for blastocysts vitrified and warmed between the two vitrification/warming kits, or between the two protocols for the Global Fast Freeze/Thaw Kits.
Conclusions
Blastocyst vitrification can be achieved with equal success using simplified protocols and cheaper and easy to load freezing straws, providing simultaneously increased safety, and efficiency with lower cost, when compared with vitrification using specialized embryo vitrification devices.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vitrification is a modern method for cryopreservation of human embryos, currently available in most IVF centers. The ease of vitrification, its reduced procedure time and the published success rates make it the preferred method for storing human oocytes and embryos [1–3]. Several cryoprotectant kits and carriers are available in the market, but some require extensive training, and others have been shown to result in inconsistent survival rates following thawing [4–6]. Dimethyl sulphoxide (DMSO) is the most frequently used cryoprotectant, due to its rapid transport through cell membrane and it has been shown to be efficient for oocyte, embryo and blastocyst vitrification. However, the possible toxicity of DMSO is of concern [7], the minute devices required are expensive and difficult to handle, and the timing of exposures to the various solutions must be extremely accurate. Furthermore, there is increasing concern with open vitrification systems, due to the stricter regulations adopted by different countries in order to minimize the risk of cross-contamination. Some vitrification kits combine DMSO with another cryoprotectant, in order to reduce the toxicity of the vitrification solutions available (e.g. Vit Kit, Irvine Scientific, CA; [8]). In fact, the first pregnancy and birth from the transfer of vitrified blastocysts were achieved using EG and DMSO as cryoprotectants [9, 10]. This combination still requires small and expensive devices for vitrification to occur. Others formulations are DMSO-free: some still employ small devices and require very small vitrification volumes (eg. Rapid-I™ kit, Vitro Life, Sweden, [11]), while others are volume independent, use larger straws which are easier to handle and allow for longer embryo exposure to vitrification solutions (e.g. Global Fast Freeze kit, LifeGlobal, LLC, Canada, [12, 13]).
The assessment of different vitrification systems is frequently done using animal embryos [14–17], discarded human embryos [18, 19] or embryos donated for research purposes [20]. These studies commonly included evaluation of survival following vitrification/warming, subsequent development and re-expansion [17, 20]. For protocols involving slower cooling rates, it is important to assess cell damage after vitrification because of the possibility that lower cooling rates may lead to ice crystal formation both inside and outside the blastocyst and thereby cause cell death. Cell damage, assessed in terms of DNA damage, was found to be similar between embryos vitrified with open and closed carriers [17]. Similarly, a recent report showed no difference in terms of percentage of dead cells, between open and closed vitrification systems, where cooling rates were slower [20].
In the present study, we evaluated and compared survival, re-expansion, and percentage of live cells following vitrification and warming of Day 5 and 6 human blastocysts, vitrified and warmed with the Vit Kit Freeze/Thaw (Irvine Scientific, CA), or with two protocols using the Global Fast Freeze/Thaw Kits (LifeGlobal, Canada). Furthermore, cell survival of the vitrified/thawed embryos was compared with non-vitrified controls.
Material and methods
Ethical approval
The study was approved by the Ethical Committee of the University Hospital of Antwerp, Belgium (registration number: B300201212114 of 02/7/2012) and a written informed consent was obtained from all patients participating in the study. A total of 764 frozen Days 2 and 3 embryos from 191 patients of all age groups, which had undergone the same freezing protocol in the same clinic, were donated for scientific research, thawed and cultured until day 5–6. In addition, 208 discarded embryos (1 or 3 PN embryos, biopsied & rejected embryos or too fast/too slow embryos) from 61 patients were cultured until Day 5–6.
Thawing procedure and culture conditions
Frozen Days 2 and 3 embryos were thawed using a commercially available thawing media (Embryo Thawing Pack, Origio, Denmark). Frozen straws were thawed 50 s in air and their contents expelled into a Petri dish. The cryoprotectant was then removed at room temperature by a step-wise dilution, according to the manufacturer’s protocol. All embryos were cultured until Day 5 or Day 6 in 20 μl drops of Global Total media (LifeGlobal) under mineral oil at 37 °C in an atmosphere of 5 % CO2 in air.
Experimental design
As shown in Table 1, a total of 154 blastocysts from 96 patients were randomly allocated into 4 treatment groups. Group 1 blastocysts (27 Day 5 embryos, of which 6 were biopsied and carried specific gene mutations, and 2 Day 6) were vitrified using the Vit Kit. Group 2 blastocysts (13 Day 5, 34 Day 6), and Group 3 blastocysts (30 Day 5, 18 Day 6) were vitrified using the Global Fast Freeze Kit, with a direct plunge, or a holding period at −100 °C, respectively. Group 4 (control) blastocysts (6 Day 5, 24 Day 6) were not vitrified. Embryos from Groups 1, 2 and 3 were vitrified/thawed according to the protocols described below and then cultured for a period of 24 h. Control blastocysts (n = 30) and blastocysts surviving vitrifications/warming and culture to 24 h, (n = 93) were stained to determine the number of live and dead cells. Of these, blastocysts that were contracted after staining (n = 6), lost during the staining procedure (n = 4), or with poor staining results (n = 4) were not included in the cell survival analysis.
Embryo quality assessment and digital imaging
Assessment of blastocyst morphological quality and stage (Days 5 or 6) was done according to the classification developed by Gardner & Schoolcraft [21]. Briefly, embryos were classified according to stage as: 1–the blastocoelic cavity represented less than half of the volume of the embryo, 2–the blastocoelic cavity was more than half of the volume of the embryo, 3–full blastocyst, cavity completely filled the embryo, 4–expanded blastocyst, cavity was larger than the embryo with thinning of the zona pellucida, 5–hatching out of the zona pellucida, or 6–hatched out of the zona pellucida. The inner-cell mass was classified as: A–many cells, tightly packed, B–several cells, loosely grouped, or C–very few cells. The trophectoderm was classified as: A–many cells forming a cohesive layer, B–few cells forming a loose epithelium, C–very few large cells. Assessment of stage and morphology was done before vitrification and after warming and re-expansion was assessed following overnight culture.
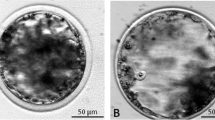
Digital images of each blastocyst were acquired before vitrification, immediately after thawing, and after 24 h culture, using a digital still camera (GC-X3E; JVL, Yokohama, Japan) mounted on an inverted optical microscope (TDM, Nikon), with a thermal control microscope stage (CO 102; Linkam Scientific Instruments Ltd, Tadworth, Surrey, UK) (Fig. 1).

Images of human Days 5 and 6 blastocyts before vitification (a, b, c) and after vitrification and waming with the Vit-Kit–direct plunge protocol (d), the Fast Freeze–direct plunge protocol (e) and the Fast Freeze −100 °C step protocol (f). After 24 h in culture, all corresponding blastocysts had re-expanded (g, h, i). Respective fluorescent live/dead stained human blastocysts 24 h after warming and culture (j, k, l). Cells with blue nuclei were intact and cells with pink/red nuclei were lysed
Protocol for vitrification and warming blastocysts
Group 1 blastocysts were vitrified using the Vit Kit Freeze (Irvine Scientific), which comprises 2 solutions. The equilibration solution, contains DMSO and ethylene glycol and the vitrification solution, contains DMSO, ethylene glycol, and sucrose. The basal medium for these solutions is modified M199 buffered media, supplemented with dextran serum supplement (DSS) and gentamicin. The method of blastocyst vitrification was as described by Kuwayama et al. [8], with slight modifications and HSV straws were used. All procedures were done at room temperature. Briefly, blastocysts were placed into the equilibration solution, for 5–15 min, until shrinking and re-expansion was observed. The blastocysts were then placed in 3 consecutive drops of vitrification solution for 5, 5, and 10 s, respectively. The blastocysts were then aspirated from the last vitrification drop using a micropipette and deposited in the gutter of the capillary rod (Cryo Bio System), in a drop of less than 1 μl, at 1 mm from the end. The capillary rod was placed into a HSV straw (Cryo Bio System) and the straw sealed. The HSV straw was then plunged directly into liquid nitrogen.
Group 1 blastocysts were thawed using the Vit Kit Thaw (Irvine Scientific) which comprises 3 solutions containing decreasing concentrations of sucrose. Thawing was done by plunging the previously opened straws into the thawing solution held at 37 °C in a petri dish, and by leaving the blastocyst in this solution for 1 min. Subsequently, blastocysts were transferred to a dilution solution for 4 min, and to 2 drops of washing solutions, for 4 min each.
Group 2 and 3 blastocysts were vitrified using the Global Fast Freeze Kit (LifeGlobal LLC, Guilford, CT, USA) which comprises 3 vitrification solutions, containing a combination of glycerol, ethylene glycol, human serum albumin (HSA), HEPES, and the base components of Global medium. Briefly, blastocysts were transferred to a drop of vitrification solution 1, where they were held for 5 min and subsequently to vitrification solution 2 where they were kept for another 5 min, at room temperature. The blastocyst was then transferred to a drop of vitrification solution 3 and immediately loaded into a 0.30 ml previously labeled freezing straw (CBS High security straw, Cryo Bio System, IMV Technologies, France), attached to a straw holding and aspiration device. The blastocyst was kept in a column of vitrification solution 3, between 2 air bubbles. The straw was then sealed with an appropriate heat sealer. For Group 2 blastocysts, the straws were then plunged directly into liquid nitrogen. For Group 3 blastocysts, the straws were held vertically together with a thermocouple probe, and lowered into the mouth of a liquid nitrogen tank. Once the temperature reached −100 °C, the straw was held still for 3 min, before it was plunged into liquid nitrogen.
Group 2 and 3 blastocysts were thawed using the Global Fast Freeze Thaw Kit (LifeGlobal) which comprises 5 solutions, with decreasing concentrations of sucrose. Briefly, the straw was held in air for 5 s and then in a 20 °C water bath for 10 s. One end of the straw was cut off and the blastocysts recovered into thawing solution 1. After 5 min, the blastocysts were transferred consecutively to thawing solutions 2, 3 4 and 5, where they were held for 5 min each. All procedures were done at room temperature, except the last step where blastocysts were placed in the thawing solution 5 and the dish was moved to a warm surface, at 37 °C.
After thawing, all blastocysts were transferred to a pre-equilibrated dish containing 20 μl culture drops of Global Total medium (LifeGlobal) under mineral oil and incubated overnight at 37 °C in standard incubation conditions (5 % CO2 in air) in order to assess re-expansion and post-warming survival.
Survival assessment by vital staining
Following 24 h of culture after thawing, embryos were stained individually to determine the number of live and dead cells. Blastocysts were stained with propidium iodide (PI) for staining dead cells (Life-dead cell staining kit; Biovision, CA, US) and Hoechst stain 33342 (Thermo scientific, IL, USA) for staining the nucleus of the fixed cells, as described by Stachecki and collegues [12], with slight modifications. Briefly, blastocysts were incubated in 400 μl of staining buffer medium with 1 μl of PI (2.5 mg/ml), in the dark, at 37 °C for 15 min, washed in buffer medium, fixed individually in 70 % ethanol at 5 °C for 5 min, and incubated in 400 μl of 70 % ethanol containing 1 μl of Hoechst stain 33342 (12.3 mg/ml) at room temperature for 10 min. The embryos were subsequently loaded individually into a 10 μl drop of mounting media (VectaShield, Vector Laboratories, Burlingame, CA) on a clean microscope slide. A cover slip was fixed to the slide using high vacuum grease (Down corning, US) as a spacer and the blastocyst was imaged immediately using a fluorescence beam (Olympus U-RFL-T beam) mounted on an inverted microscope (Olympus BX51; Tokyo, Japan) to determine total cell number and the ratio of live-dead cells in each blastocyst. The nucleii of the dead blastomeres stained red and the nucleus of live cells (membrane intact) stained blue. Digital images of the fluorescently labeled blastocysts were acquired using a digital still camera (Olympus, E-450) mounted on a fluorescence microscope. Cell number was assessed with a Java-based image processing program (ImageJ; National Institute of Health) and the percentage of live cells calculated for each blastocyst.
Statistical analysis
The effect of vitrification treatment on blastocyst survival immediately after thawing, and survival and expansion at 24 h after thawing was evaluated by Pearson Chi-square analysis. The effect of vitrification treatment and day of vitrification on percentage of live cells following vitrification/warming were analyzed using a General Linear Model. The two protocols of Fast Freeze (Groups 2 and Groups 3) were also compared for blastocyst survival and expansion using Pearson Chi-square analysis, and the percentage of live cells using a General Linear Model. The combined results for Groups 2 and 3 (Fast Freeze) were compared with Group 1 (Vit Kit) for blastocyst survival and expansion by Pearson Chi-square analysis, and for percentage of live cells by a General Linear Model.
Results
Representative digital images of blastocysts before and after vitrification using the Vit Kit and Fast Freeze kits are shown in Fig. 1 and the distribution of embryos by stage and treatment is described in Table 1.
As shown in Fig. 2, there were no overall effects of vitrification treatment (Group 1, Vit Kit direct plunge, Group 2, Fast Freeze-direct plunge; Group 3, Fast Freeze −100 °C step) on immediate survival (P = 0.281), or survival (P = 0.895) or expansion (P = 0.481) after 24 h of culture following thawing. There were also no effects of vitrification treatment (including Group 4, controls, P = 0.648), day of blastocyst appearance (P = 0.615) or treatment X day interaction (P = 8.810) on percentage of live cells (Table 2). Comparison of the two vitrification protocols for Fast Freeze (Group 2 vs Group 3) showed no effect on immediate survival (P = 0.117), survival (P = 0.766) or expansion (P = 0.883) after 24 h of culture following thawing, or percentage of live cells (P = 0.583).

Blastocyst survival and expansion following vitrification and warming. The numbers in parentheses are the number of blastocysts within each category. There was no significant effect of vitrification treatment on immediate survival (P = 0.117), survival after 24 h (P = 0.766), or expansion after 24 h (P = 0.833)
Group 1 (Vit Kit–Direct plunge) was skewed towards stage-1 embryos (Table 1). Consequently, a comparison was made between stage-1 blastocysts vitrified/thawed using the Vit Kit (n = 11), and stage-1 blastocysts vitrified/thawed using the Global Fast Freeze kit (combined Groups 2 and 3, n = 14). As noted above, there were no significant differences between Groups 2 and 3 for survival at thaw, survival after 24 h, expansion after 24 h, or percentage of live cells, and it was therefore valid to combine the results for Groups 2 and 3. As shown in Fig. 3, immediate survival and survival after 24 h culture was not different between the vitrification treatments, but there was a tendency (P = 0.080) for expansion after 24 h culture to be greater for blastocysts vitrified using the Global Fast Freeze kit. When only stage-1 blastocysts were compared, there was no effect of vitrification treatment on the overall percentage of live cells, but the mean total number of cells for Group 1 (Vit Kit - direct plunge) was significantly lower than that of Groups 2 and 3 combined (Table 3).

Survival and expansion of stage-1 blastocysts following vitrification and warming. * P = 0.102, ♦ P = 0.840, ∇ P = 0.080
Discussion
Cell survival was not different between vitrified and non-vitrified blastocysts, thus vitrification did not affect cell survival in any of the treatment groups. These findings confirm earlier results, which showed that warmed blastocysts that survived vitrification were not different from fresh blastocysts, in terms of quality, DNA and chromosome integrity, ultrastructure, and developmental competence [11, 17, 22–25].
Immediate survival, survival and re-expansion after 24 h, and percentage of live cells were not different for blastocysts vitrified and warmed between the two vitrification/warming kits. This was observed despite the fact that 6 Day 5 embryos of Group 1 (Vit kit–direct plunge) were biopsied on Day 3, which could have negatively affected their development and impaired cell health. Furthermore, the distinct developmental stages of the embryos in different groups did not affect overall cell survival. Overall, these results confirmed that when combined with ethylene glycol, both DMSO, with its fast penetrating characteristics and low molecular weight, and glycerol, with higher molecular weight but moving across the plasma membrane predominantly by facilitated diffusion through aquaporins 3 [26], are efficient cryoprotectants for blastocyst vitrification, as earlier demonstrated by Stachecki et al. [12, 13]. Thus, blastocyst vitrification could be effectively achieved with larger carriers, which were sealed to avoid cross contamination during long term storage, using longer loading and exposure times and without the need of blastocoel collapse. Slower cooling rates were achieved with the Fast Freeze–direct plunge and with the Fast Freeze −100 °C step when compared with the Vit Kit–direct plunge group, but high and reproducible pregnancy rates have been reported using this method of vitrification (mean pregnancy rate of 65 %, according to Stachecki et al. [12]).
Because Group 1 was skewed towards Stage-1 blastocysts, only embryos at this developmental stage were compared regarding cell number. It was found that the total number of cells was significantly lower for stage-1 embryos of Group 1. The possible explanation for this observation might be related to the fact that all stage 1 embryos were vitrified on Day 5 in Group 1 and 1 embryo of the same group was submitted to biopsy on Day 3, losing 2 cells during this procedure. Nonetheless, the number of stage-1 blastocysts was considerably low for both groups (10 for Groups 2 and 3 combined and 8 for Group 1–direct plunge).
Neither survival, nor percentage of live cells, was significantly different between the two protocols of Global Fast Freeze/Thaw Kits. The results obtained with the Fast Freeze direct plunge were in accordance with those observed in a preliminary study where a small sample of surplus human blastocysts donated for research were vitrified with the S3 method (basis of the Global Fast Freeze media, using a protocol similar to that used in the Fast Freeze-direct plunge group) and subsequently stained, leading to a survival rate following warming of 84 % and a cell survival rate of 87 % [12]. Thus, we could document and confirm that the simplified version of the Fast Freeze protocol, including the direct plunge of the freezing straw into liquid nitrogen (Fast Freeze-direct plunge), leads to equivalent results to those obtained with the original protocol version (S3 vitrification system; [13]). Furthermore, re-expansion rate, which is a positive prognostic marker associated with significantly increased implantation and clinical pregnancies [27] was similar for both protocols, suggesting that clinical outcomes will not be significantly different.
One limitation of this study was the skew towards stage-1 blastocysts in Group 1. It would be interesting to investigate if an equal distribution of blastocysts through all developmental stages in all groups would lead to similar survival rates following vitrification, warming and culture. Results could have also been affected by the source of embryos, either frozen/thawed or fresh and discarded. Thereby, using the same source of embryos would have removed this potentially confounding effect. Finally, another weakness of the study was the reduced number of stained blastocysts. It is possible that a larger number of stained blastocysts would help clarifying differences in total cell number observed between Groups 1, 2 and 3.
In conclusion, blastocyst vitrification can be performed with equal degree of success utilizing simplified protocols and cheaper tools, without affecting the safety and efficiency of the procedure, when compared to other more complex vitrification devices.
References
Loutradi KE, Kolibianakis EM, Venetis CA, Papanikolaou EG, Pados G, Bontis I, et al. Cryopreservation of human embryos by vitrification or slow freezing: a systematic review and meta-analysis. Fertil Steril. 2008;90:186–93.
Kolibianakis EM, Venetis CA, Tarlatzis BC. Cryopreservation of human embryos by vitrification or slow freezing: which one is better? Curr Opin Obstet Gynecol. 2009;21:270–4.
AbdelHafez FF, Desai N, Abou-Setta AM, Falcone T, Goldfarb J. Slow freezing, vitrification and ultra-rapid freezing of human embryos: a systematic review and meta-analysis. Reprod Biomed Online. 2010;20:209–22.
Antinori M, Licata E, Dani G, Cerusico F, Versaci C, Antinori S. Cryotop vitrification of human oocytes results in high survival rate and healthy deliveries. Reprod Biomed Online. 2007;14:72–9.
Desai N, Blackmon H, Szeptycki J, Goldfarb J. Cryoloop vitrification of human day 3 cleavage-stage embryos: post-vitrification development, pregnancy outcomes and live births. Reprod Biomed Online. 2007;14:208–13.
Matsunari H, Maehara M, Nakano K, Ikezawa Y, Hagiwara Y, Sasayama N, et al. Hollow fiber vitrification: a novel method for vitrifying multiple embryos in a single device. J Reprod Dev. 2012;58:599–608.
Ali J, Shelton JN. Design of vitrification solutions for the cryopreservation of embryos. J Reprod Fertil. 1993;99:471–7.
Kuwayama M, Vajta G, Ieda S, Kato O. Comparison of open and closed methods for vitrification of human embryos and the elimination of potential contamination. Reprod Biomed Online. 2005;11:608–11.
Yokota Y, Sato S, Yokota M, Ishikawa Y, Makita M, Asada T, et al. Successful pregnancy following blastocyst vitrification: case report. Hum Reprod. 2000;15:1802–3.
Yokota Y, Sato S, Yokota M, Yokota H, Araki Y. Birth of a healthy baby following vitrification of human blastocysts. Fertil Steril. 2001;75:1027–9.
Desai NN, Goldberg JM, Austin C, Falcone T. The new Rapid-i carrier is an effective system for human embryo vitrification at both the blastocyst and cleavage stage. Reprod Biol Endocrinol. 2013;11:41.
Stachecki JJ, Garrisi J, Sabino S, Caetano JP, Wiemer KE, Cohen J. A new safe, simple and successful vitrification method for bovine and human blastocysts. Reprod Biomed Online. 2008;17:360–7.
Stachecki JJ, Cohen J. S3 vitrification system: a novel approach to blastocyst freezing. J Clin Embryol. 2008;11:5–14.
Nedambale TL, Dinnyés A, Yang X, Tian XC. Bovine blastocyst development in vitro: timing, sex, and viability following vitrification. Biol Reprod. 2004;71:1671–6.
Valojerdi MR, Salehnia M. Developmental potential and ultrastructural injuries of metaphase II (MII) mouse oocytes after slow freezing or vitrification. J Assist Reprod Genet. 2005;22:119–27.
Larman MG, Gardner DK. Vitrification of mouse embryos with super-cooled air. Fertil Steril. 2011;95:1462–6.
AbdelHafez F, Xu J, Goldberg J, Desai N. Vitrification in open and closed carriers at different cell stages: assessment of embryo survival, development. DNA integrity and stability during vapor phase storage for transport. BMC Biotechnol. 2011;11:29.
Cremades N, Sousa M, Silva J, Viana P, Sousa S, Oliveira C, et al. Experimental vitrification of human compacted morulae and early blastocysts using fine diameter plastic micropipettes. Hum Reprod. 2004;19:300–5.
Zhang X, Trokoudes KM, Pavlides C. Vitrification of biopsied embryos at cleavage, morula and blastocyst stage. Reprod Biomed Online. 2009;19:526–31.
Hashimoto S, Amo A, Hama S, Ohsumi K, Nakaoka Y, Morimoto Y. A closed system supports the developmental competence of human embryos after vitrification: closed vitrification of human embryos. J Assist Reprod Genet. 2013;30:371–6.
Gardner DK, Schoolcraft WB. In vitro culture of human blastocyst. In: Jansen R, Mortimer D, editors. Towards reproductive certainty: infertility and genetics beyond. Carnforth: Parthenon Press; 1999. p. 378–88.
Escribá MJ, Escobedo-Lucea C, Mercader A, de los Santos MJ, Pellicer A, Remohí J. Ultrastructure of preimplantation genetic diagnosis-derived human blastocysts grown in a co-culture system after vitrification. Fertil Steril. 2006;86:664–71.
Ku PY, Lee RK, Lin SY, Lin MH, Hwu YM. Comparison of the clinical outcomes between fresh blastocyst and vitrified-thawed blastocyst transfer. J Assist Reprod Genet. 2012;29:1353–6.
Chatzimeletiou K, Morrison EE, Panagiotidis Y, Vanderzwalmen P, Prapas N, Prapas Y, et al. Cytoskeletal analysis of human blastocysts by confocal laser scanning microscopy following vitrification. Hum Reprod. 2012;27:106–13.
Muthukumar K, Kamath MS, Mangalaraj AM, Aleyamma T, Chandy A, George K. Comparison of clinical outcomes following vitrified warmed day 5/6 blastocyst transfers using solid surface methodology with fresh blastocyst transfers. J Hum Reprod Sci. 2013;6:59–64.
Kasai M, Edashige K. Movement of water and cryoprotectants in mouse oocytes and embryos at different stages: relevance to cryopreservation. In: Chian R, Quinn P, editors. Fertility cryopreservation. New York: Cambridge University Press; 2010. p. 16–23.
Ebner T, Vanderzwalmen P, Shebl O, Urdl W, Moser M, Zech NH, et al. Morphology of vitrified/warmed day-5 embryos predicts rates of implantation, pregnancy and live birth. Reprod Biomed Online. 2009;19:72–8.
Acknowledgments
The authors would like to thank Dr. Don Rieger, LifeGlobal LLC, Guelph, ON, Canada for his critical review of the manuscript.
Conflict of interest
Global Fast Freeze/Thaw kits were provided by IVFonline. ASL is a technical consultant for IVFOnline.
Author information
Authors and Affiliations
Corresponding author
Additional information
Capsule Survival, re-expansion, and percentage of live cells were not different for blastocysts vitrified between two vitrification/warming kits and therefore vitrification can be performed with equal success utilizing simplified protocols and cheaper tools.
Rights and permissions
About this article

Cite this article
Lopes, A.S., Frederickx, V., Van Kerkhoven, G. et al. Survival, re-expansion and cell survival of human blastocysts following vitrification and warming using two vitrification systems. J Assist Reprod Genet 32, 83–90 (2015). https://doi.org/10.1007/s10815-014-0373-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-014-0373-2