
Abstract
Purpose
To research the association between the single nucleotide polymorphisms (SNPs) of three spermatogenesis-related genes (USF1, GTF2A1L and OR2W3) and non-obstruction azoospermia (NOA).
Methods
We investigated 361 NOA cases and 368 controls from the Chinese Han population, and we used Sequenom iplex technology to analyze the candidate 9 SNPs from the USF1, GTF2A1L and OR2W3 genes.
Results
In this study, we found that the variant rs2516838 of USF1 was associated with NOA susceptibility (P = 0.020, OR = 1.436), and the haplotype TCG of the variants rs1556259, rs2516838, and rs2774276 of USF1 conferred an increased risk of NOA (P = 0.019, OR = 1.436). Furthermore, we found that the rs11204546 genotype of OR2W3 and the rs11677854 genotype of GTF2A1L were correlated with the FSH level in the patients (P = 0.004 and P = 0.018, respectively).
Conclusions
Our results provided a new insight into susceptibility of USF1 variant with male infertility. Clinically, the SNPs (rs11204546 of OR2W3 and rs11677854 of GTF2A1L ) might be additional valuable molecular predictive markers for assessing the treatment of NOA patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Approximately one out of six couples experiences infertility and nearly half of these cases can be ascribed to the male partner [1]. Regardless of various causative factors such as varicocele, obstruction of spermatic ducts, agglutination of sperm, impotence and hormonal imbalance, a significant proportion of male infertility attributes to idiopathic factor [2, 3]. It is demonstrated that single nucleotide polymorphisms (SNPs) in genes are associated with a variety of clinical subtypes of male infertility, including non-obstructive azoospermia (NOA), which is clinically characterized by the absence of sperm in the testis as a result of impaired spermatogenesis [4–7].
During spermatogenesis, gene expressions are regulated at the transcriptional level to ensure the differentiation of germ cells [8]. Upstream stimulatory factor (USF) is a ubiquitously expressed cellular transcription factor. Purified USF consists of 2 related polypeptides, that is USF1 and USF2. Upstream transcription factor 1 gene (USF1) locates in human chromosome 1q23.3. In terms of the structure of USF1 protein, USF1 contains a C-terminal DNA-binding domain that includes a helix-loop-helix motif and a leucine repeat which is required for efficient DNA binding and USF1 dimerization [9]. USF1 is one of the transcriptional factors of the spermatogenesis-related genes. There were two mechanisms elaborating how the USF1 gene influenced the spermatogenesis-related genes. One acted through controlling the number of Sertoli cells. In the adult testis, Sertoli cells are essential to provide the physical environment for germ cells to develop mature sperms. The number of germ cells that could be supported depended on the duration of the proliferation period in Sertoli cells [10]. Therefore, timing Sertoli cell differentiation was critical for determining the final number of Sertoli cells and the upper limit of male fertility. The USFs (USF1 and USF2) regulated numerous Sertoli cell genes which were indispensable during differentiation [11, 12], and the other functioned through initiating the spermiogenesis. A previous study showed that the transcription factor USF specially bound and activated the Miwi promoter in mice [13]. Miwi, a cytoplasmic protein, was specifically expressed in the testis from pachytene spermatocytes to round spermatids and was required for initiating spermiogenesis [14, 15]. USF controlled Miwi expression through methylation-mediated regulation. The CpG islands in the Miwi promoter were unmethylated in the pachytene spermatocytes and round spermatids where Miwi was expressed. If the CpG island within the E2 box to which the USFs bind was methylated, the binding of the USF would be inhibited, thereby Miwi expression would not occur [13]. In that case, the Miwi-dependent initiating spermiogenesis would not take place.
In this study, we hypothesized that USF1, GTF2A1L and OR2W3 might be potential candidates for the disruption of spermatogenesis. We assumed that there might be genetic associations between variants of the genes and NOA susceptibility. To verify this hypothesis, we performed a genotyping analysis for 9 SNPs from USF1, GTF2A1L and OR2W3 genes in a case–control study.
Materials and methods
Ethic statement
All patients provided written informed consent for the collection of samples and subsequent analyses. This study was approved by the ethics committee for genome research of the Anhui Medical University and was conducted based on the Declaration of Helsinki Principles (reference no. 2008035, 10 January 2008).
Patients of NOA and the controls
In this study, we investigated 361 NOA patients with the age ranging from 19 to 43 and 368 controls with the age ranging from 20 to 53 from the Chinese population. Patients with NOA were collected from the Reproductive Medicine Center, the First Affiliated Hospital of Anhui Medical University, between March 2008 and July 2013. To confirm the diagnosis of azoospermia, i.e. no sperm was found after ejaculate centrifugation at 3,000 g for 10 min, we analyzed at least two semen samples. The semen analyses for sperm concentration, motility, and morphology were performed following the fifth edition of the World Health Organization manuals [16]. The NOA patients were carefully selected based on a comprehensive examination, including semen analysis, karyotype analysis, medical history, physical examination, hormone analysis and Y chromosome microdeletions screening. Patients with a history of orchitis, maldevelopment of testis, mono- or bilateral cryptorchidism, varicocele, hypogonadism, obstruction of vas deferens, chromosomal abnormalities, and Y microdeletions were excluded from this study [17, 18]. The controls were all fertility men who had fathered 1 or more healthy children. All control donors were enrolled from the same hospital where the patients were recruited. All the subjects tested were the Chinese Han population. Also, we used Electro-Chemiluminescence Immunoassay system (Roche Diagnostics, Germany) to measure the FSH level of the patients. We divided the patients into two subtypes: FSH <12.4mIU/ml and FSH ≥12.4mIU/ml, according to the normal range of FSH level (1.5–12.4mIU/ml). Furthermore, we designed 10 ml of the patient’s testis volume as a threshold on the basis of the previous study [19] . All the testis volume of the controls were larger than 10 ml.
Extraction of peripheral blood DNA
we extracted the genomic DNA from peripheral blood using QiAamp DNA Blood MiDi Kit (Qiagen Inc., Germany), according to the manufacturer’s protocol. DNA purity and concentrations were determined using a NanoDrop Spectrophotometer (ND-100, wilmington, DE) of full wave length and standardized to 30 ng/μl.
SNP selection and genotyping
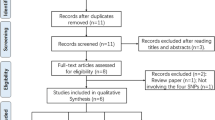
We selected 9 SNPs in this study using Haploview software [20], including 3 USF1 SNPs (rs2516838, rs1556259, rs2774276), 4 GTF2A1L SNPs (rs1916838, rs11677854, rs10432667, rs13024367) and 2 OR2W3 SNPs (rs10888267, rs11204546). All the candidate SNPs were selected from the Chinese population of International HapMap Project Web site (http://hapmap.ncbi.nlm.nih.gov/) with MAF > 0.05. Locus-specific PCR and detection primers were designed using the MassARRAY Assay Design 3.0 software (Sequenom, San Diego, California) following the manufacturer’s instructions. Then we amplified the DNA samples by multiple PCR reactions, and the PCR products were used for locus-specific single-base extension reactions. The resulting products were desalted and transferred to a 384-element SpectroCHIP array. We performed a genotyping analysis of the SNPs for fast-track validation analysis using the Sequenom MassArrsy system (Sequenom, San Diego, CA). Subsequently, we used fifteen nanograms genomic DNA to genotype each sample. We performed allele detection using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and the mass spectrograms were analyzed by the MassARRAY Typer software (Sequenom).
Statistical analysis
We used T-test to compare the clinical characteristics between the controls and cases. We determined the Hardy-Weinberg equilibrium (HWE) by the controls (P > 0.01). The HWE of the patients, the differences in frequency distributions of the genotypes between the cases and the controls and the correlations between the clinical data and all the SNPs were calculated in two ways: SHEsis software (http://analysis.bio-x.cn/myAnalysis.php) [21] and SPSS release 17.0 statistical package. Moreover, the sample powers of each SNP were calculated using PS (power and sample size calculations) software [22] and the power calculation of the SNPs detected reached the research need (>0.08). In addition, for Chi-square test, Fisher exact test was used when the number of genotype ≤ 5. In this study, of the 9 SNPs evaluated, we considered the associations with a p value of <0.05 to be of significance. We did not adjust the data for multiple comparisons.
Results
Characteristics of the study population
We summarized clinical characteristics of the NOA cases and the controls in Table 1. The results showed that the testicle volume in NOA cases was significantly smaller than that in the controls (P < 0.001, both the sides).
The polymorphisms of USF1, GTF2A1L and OR2W3 genes and risk of NOA
In this study, we investigated 3 USF1 SNPs (rs2516838 G/C, rs1556259 C/T, rs2774276 G/C), 4 GTF2A1L SNPs (rs1916838 T/C, rs11677854 C/T, rs10432667 A/G, rs13024367 C/G) and 2 OR2W3 SNPs (rs10888267 T/C, rs11204546 C/T). All of the 9 SNPs of interest did not deviate from the HWE (P > 0.01) in the controls. We found that C allele of rs2516838 was associated with NOA (P = 0.02, odds ratio [OR] =1.436, 95 % confidence interval [CI] = 1.059–1.947, Table 2). Table 3 showed genetic model of the SNP rs2516838 between the controls and the cases. We found that the individuals with CG genotype of rs2516838 increased risk susceptibility to the NOA in the codominant model (P = 0.008, OR = 1.610, 95 %CI = 1.133–2.287), and the individuals with the minor allele C of rs2516838 increased the risk susceptibility to the NOA in the dominant model (P = 0.009, OR = 1.566, 95 %CI = 1.115–2.199).
Haplotypes of USF1 gene and risk of NOA
To investigate potential combination effects of USF1 SNPs on susceptibility of spermatogenic failure further, we did a haplotype analysis in USF1 gene and found a significant haplotype associated with NOA. The haplotype TCG containing all the 3 SNPs of USF1 gene (rs1556259, rs2516838, rs2774276) exhibited significantly increased susceptibility to suffering from spermatogenic failure in the Chinese patients (P = 0.019, OR = 1.436, 95 % CI = 1.059–1.948, Table 4).
Association of the 9 polymorphisms with clinical data of NOA
We further performed a stratified association analysis of the 9 SNPs with the testis volume and the level of FSH in the patients. Our data indicated that there were significant correlations for rs11204546 of OR2W3 (C > T, P = 0.004) and rs11677854 of GTF2A1L (C > T, P = 0.018) with the FSH level in the patients, rather than the testis volume (P > 0.05) (Table 5).
Discussion
In this study, we chose 9 SNPs from USF1, GTF2A1L, OR2W3 genes to investigate the relationship between non-obstructive azoospermia and the variants. The results showed that rs2516838 of USF1 significantly led to an increased risk of spermatogenic failure. Due to the presence of the USF1/USF2a heterodimers [23], USF1 was not yet characterized alone. Evidences showed that USF1 played a pivotal role in spermatogenesis [11–13]. The potential mechanisms that the SNP rs2516838 involved in NOA remained unclear. It was supposed that 1) the variations might influence the USF1 expression. Based on the analysis performed by the GoldenPath (www.genome.ucsc.edu) [24], a database forecasting the function of SNPs in transcriptional regulation, the G to C change of rs2516838 might disturb the gene transcription. The deficient transcription of USF1 would decrease the level of USF1 protein. During proliferation, low level of USF1 expression in Sertoli cells limited the USF recruitment to E-box motifs within the promoter regions of Nr5a1 and Shbg genes which were vital for Sertoli cell differentiation [12], therefore the progress of the spermatogenesis might be repressed as a result of the aberrant differentiation of Sertoli cells, NOA would take place; 2) We found that the minor allele C of the SNP rs2284922 located in the conserved region of USF1 gene by means of the F-SNP database [25], a web-based SNP analysis tool. Such an alteration in the gene might lead to an abnormality of gene expression or weaken the stability of gene [26]; 3) the variation of rs2516838 might relate to the initiation of spermiogenesis. There were studies revealing that the rs2516838 polymorphism of USF1 was associated with methylation of genomic DNA [27, 28]. As Miwi gene was essential for initiating spermiogenesis, and a recent investigation implied that the methylation of CpG islands within the proximal promoter of Miwi gene revealed a marked correlation with the expression of Miwi [13]. We supposed that the variant rs2516838 of USF1 might influence the methylation status of Miwi gene and indirectly lead to NOA; and 4) rs2516838 might be in linkage disequilibrium with other variants in USF1 gene or other genes that could play a concurrent role in impaired spermatogenesis, as those in other complex diseases [29].
To address potential combination effects of variants of USF1 on risk of spermatogenesis defect, we performed a haplotype analysis among the 3 SNPs rs1556259, rs2516838 and rs2774276 of USF1 gene. We found that the frequency of the haplotype TCG of the three SNPs was significantly higher in the NOA patients than that in the controls (P = 0.019, OR = 1.436, and 95 % CI = 1.059–1.948). In comparison with the other 3 haplotypes (Table 4), the minor allele C of rs2516838 would probably exert a dominant force to NOA. Thus, we supposed that the Chinese individuals who carried the haplotype TCG with the minor allele C of the rs2516838 might be liable to suffering from NOA.
Furthermore, GTF2A1L, as a germ cell-specific transcription and a substitute for TF II A in male germ cells, was involved in spermatogenesis [30]. None or less expression of GTF2A1L protein in the infertile patients with spermatogenetic disturbances indicated that GTF2A1L gene might be correlated with NOA [31]. However, in this study, we failed to identify any associations between the variants of GTF2A1L gene and NOA. In addition, although two independent studies demonstrated an association of the SNP rs11204546 of OR2W3 gene with azoospermia in the European descent [32, 33], in this study we found no association of the OR2W3 rs11204546 with Chinese NOA. The reasons for the divergences among the investigations appeared unknown. It would be attributable either to the strictly controlled recruitment of our clinical cases or to the ethnic population differences. Moreover, in this study, we found that there were significant correlations for rs11204546 of OR2W3 (C > T, P = 0.004) and rs11677854 of GTF2A1L (C > T, P = 0.018) with the FSH level in the patients (Table 5). Interestingly, a previous study indicated that the FSH level might be a predictive factor to obtain elongated spermatids by testicular sperm extraction for azoospermic patients [34]. Therefore, we hypothesized these two FSH-related SNPs (rs11204546 and rs11677854) might be a valuable molecular predictive markers of sperm/elongated spermatids retrieval rate for NOA patients. But yet, additional clinical studies are needed to confirm the hypothesis.
In conclusion, our study showed that the USF1 variant rs2516838 was associated with spermatogenesis in the Chinese Han population and indicated the susceptibility of the USF1 haplotypes TCG to NOA. Clinically, we found that rs11204546 and rs11677854 were associated with the FSH. Nevertheless, further investigations on the molecular mechanisms in the USF1 , OR2W3 and GTF2A1L genes might provide better understanding of the development of NOA.
References
De Kretser DM, Baker HW. Infertility in men: recent advances and continuing controversies. J Clin Endocrinol Metab. 1999;84(10):3443–50.
Rucker GB, Mielnik A, King P, Goldstein M, Schlegel PN. Preoperative screening for genetic abnormalities in men with nonobstructive azoospermia before testicular sperm extraction. J Urol. 1998;160(6 Pt 1):2068–71.
Cram DS, O’Bryan MK, de Kretser DM. Male infertility genetics—the future. J Androl. 2001;22(5):738–46.
Tüttelmann F, Rajpert-De Meyts E, Nieschlag E, Simoni M. Gene polymorphisms and male infertility–a meta-analysis and literature review. Reprod Biomed Online. 2007;15(6):643–58.
He XJ, Ruan J, Du WD, Chen G, Zhou Y, Xu S, et al. PRM1 variant rs35576928 (Arg > Ser) is associated with defective spermatogenesis in the Chinese Han population. Reprod Biomed Online. 2012;25(6):627–34.
Ruan J, He XJ, Du WD, Chen G, Zhou Y, Xu S, et al. Genetic variants in TEX15 gene conferred susceptibility to spermatogenic failure in the Chinese Han population. Reprod Sci. 2012;19(11):1190–96.
Jinam TA, Nakaoka H, Hosomichi K, Mitsunaga S, Okada H, Tanaka A, et al. HLA-DPB1*04:01 allele is associated with non-obstructive azoospermia in Japanese patients. Hum Genet. 2013;132(12):1405–11.
Tanaka H, Baba T. Gene expression in spermiogenesis. Cell Mol Life Sci. 2005;62(3):344–54.
Gregor PD, Sawadogo M, Roeder RG. The adenovirus major late transcription factor USF is a member of the helix-loop-helix group of regulatory proteins and binds to DNA as a dimer. Genes Dev. 1990;4(10):1730–40.
Sharpe RM, McKinnell C, Kivlin C, Fisher JS. Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reprod. 2003;125(6):769–84.
Wood MA, Walker WH. USF1/2 transcription factor DNA-binding activity is induced during rat Sertoli cell differentiation. Biol Reprod. 2009;80(1):24–33.
Wood MA, Mukherjee P, Toocheck CA, Walker WH. Upstream stimulatory factor induces Nr5a1 and Shbg gene expression during the onset of rat Sertoli cell differentiation. Biol Reprod. 2011;85(5):965–76.
Hou Y, Yuan J, Zhou X, Fu X, Cheng H, Zhou R. DNA demethylation and USF regulate the meiosis-specific expression of the mouse Miwi. PLoS Genet. 2012;8(5):e1002716.
Deng W, Lin H. miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev Cell. 2002;2(6):819–30.
Kuramochi-Miyagawa S, Kimura T, Yomogida K, Kuroiwa A, Tadokoro Y, Fujita Y, et al. Two mouse piwi-related genes: miwi and mili. Mech Dev. 2001;108(1–2):121–33.
Cooper TG, Noonan E, von Eckardstein S, Auger J, Baker HW, Behre HM, et al. World health organization reference values for human semen characteristics. Hum Reprod Update. 2010;16(3):231–45.
Wu B, Lu NX, Xia YK, Gu AH, Lu CC, Wang W, et al. A frequent Y chromosome b2/b3 subdeletion shows strong association with male infertility in Han-Chinese population. Hum Reprod. 2007;22(4):1107–13.
Lu C, Zhang J, Li Y, Xia Y, Zhang F, Wu B, et al. The b2/b3 subdeletion shows higher risk of spermatogenic failure and higher frequency of complete AZFc deletion than the gr/gr subdeletion in a Chinese population. Hum Mol Genet. 2009;18(6):1122–30.
Hibi H, Ohori T, Yamada Y, Honda N, Asada Y. Probability of sperm recovery in non-obstructive azoospermic patients presenting with testes volume less than 10 ml/FSH level exceeding 20 mIU/ml. Arch Androl. 2005;51:225–31.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinforma. 2005;21(2):263–5.
Shi YY, He L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005;15(2):97–8.
Dupont WD, Plummer Jr WD. Power and sample size calculations for studies involving linear regression. Control Clin Trials. 1998;19(6):589–601.
Viollet B, Lefrançois-Martinez AM, Henrion A, Kahn A, Raymondjean M, Martinez A. Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J Biol Chem. 1996;271(3):1405–15.
Tourmen Y, Baris O, Dessen P, Jacques C, Malthièry Y, Reynier P. Structure and chromosomal distribution of human mitochondrial pseudogenes. Genom. 2002;80(1):71–7.
Lee PH, Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36(Database issue):D820–4.
Venkatesh S, Kumar R, Deka D, Deecaraman M, Dada R. Analysis of sperm nuclear protein gene polymorphisms and DNA integrity in infertile men. Syst Biol Reprod Med. 2011;57:124–32.
Collings A, Höyssä S, Fan M, Kähönen M, Hutri-Kähönen N, Marniemi J, et al. Allelic variants of upstream transcription factor 1 associate with carotid artery intima-media thickness: the cardiovascular risk in young Finns study. Circ J. 2008;72(7):1158–64.
Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One. 2010;5(3):e9692.
Tan EK, Chan DK, Ng PW, Woo J, Teo YY, Tang K, et al. Effect of MDR1 haplotype on risk of Parkinson disease. Arch Neurol. 2005;62:460–4.
Ozer J, Moore PA, Lieberman PM. A testis-specific transcription factor IIA (TFIIAtau) stimulates TATA-binding protein-DNA binding and transcription activation. J Biol Chem. 2000;275(1):122–8.
Huang M, Wang H, Li J, Zhou Z, Du Y, Lin M, et al. Involvement of ALF in human spermatogenesis and male infertility. Int J Mol Med. 2006;17(4):599–604.
Aston KI, Krausz C, Laface I, Ruiz-Castané E, Carrell DT. Evaluation of 172 candidate polymorphisms for association with oligozoospermia or azoospermia in a large cohort of men of European descent. Hum Reprod. 2010;25(6):1383–97.
Plaseski T, Noveski P, Popeska Z, Efremov GD, Plaseska-Karanfilska D. Association study of single-nucleotide polymorphisms in FASLG, JMJDIA, LOC203413, TEX15, BRDT, OR2W3, INSR, and TAS2R38 genes with male infertility. J Androl. 2012;33(4):675–83.
Zitzmann M, Nordhoff V, von Schönfeld V, Nordsiek-Mengede A, Kliesch S, Schüring AN, et al. Elevated follicle-stimulating hormone levels and the chances for azoospermic men to become fathers after retrieval of elongated spermatids from cryopreserved testicular tissue. Fertil Steril. 2006;86:339–47.
Acknowledgments
We were grateful to the participants: the patients, the clinicians and the co-authors in this study.
Declaration of interests
The authors declared no potential conflicts of interest in terms of the research, authorship, and/or publication of this article. The authors alone were responsible for the content and writing of the paper.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Capsule USF1 variant rs2516838 might confer risk to NOA.
Yan Zhang and Xiao-Jin He contributed equally to this manuscript.
Rights and permissions
About this article

Cite this article
Zhang, Y., He, XJ., Song, B. et al. Association of single nucleotide polymorphisms in the USF1, GTF2A1L and OR2W3 genes with non-obstructive azoospermia in the Chinese population. J Assist Reprod Genet 32, 95–101 (2015). https://doi.org/10.1007/s10815-014-0369-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-014-0369-y