
Abstract
Objective
The aim of this study was to evaluate the impact of vitrification on mitochondrial membrane potential (ΔΨm) in human metaphase II (MII) oocytes, and the changes of ΔΨm on thawed MII oocytes.
Methods
MII oocytes were obtained from clinical IVF cycles when the oocytes were failed to fertilization within 24 h after insemination. All oocytes were randomly divided into 4 groups: non-frozen (fresh group), cultured for 0 h (0 h group), 2 h (2 h group) and 4 h (4 h group) after vitrification/thawing. All oocytes were stained with the ΔΨm-specific probe JC-1 and detected by laser scanning confocal microscope (LSCM) for mitochondrial analysis.
Results
The ΔΨm of oocytes was significantly decreased in 0 h and 2 h groups when compared with fresh group (0.93, 1.09 vs 1.34, P < 0.05), but similar between 4 h group and fresh group (1.30 vs 1.34, P > 0.05).
Conclusion
In the vitrification/thawing process, the ΔΨm of MII oocytes could have temporally dynamic changes within 2 h after thawing but would be fully recovered after 4 h culture.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the application of controlled ovarian stimulation treatment in human in vitro fertilization (IVF), oocyte cryopreservation has been regarded as an ideal option to capitalize the reproductive potential of surplus oocytes and preserve female fertility. Oocyte cryopreservation may avoid ethical and legal issues associated with human embryo freezing and provide future fertility for women who would destine to lose gonadal functions due to extra therapy, radiation or chemotherapy, and for those who wish to delay their maternity. However, for almost two decades the endeavor to preserve oocytes was frustrated. Since the poor implantation and pregnancy rate severely limited the clinical application of oocytes cryopreservation, the embryos cryopreservation has developed as a preferred choice of fertility preservation, though they have the similar survival and fertilization rate [1–3]. In recent years, vitrification cryopreservation developed rapidly, and many clinical reviews supported that the oocytes from vitrification/thawing cycles could result in similar clinical outcomes with fresh oocytes [4–7]. However, there is still a lack of basic research on the potential injury of oocytes after vitrification.
Mitochondria are specialized organelles that catalyze the formation of adenosine trisphosphate (ATP), which provides most of the energy for fertilization and embryonic development. Mitochondrial membrane potential (ΔΨm) is the foundation of energy metabolism in cells. It is a sensitive index of cell damage because it is easily to be influenced by the environment [8]. JC-1 is an ideal probe to detect ΔΨm, whose emission of red and green fluorescence only relies on ΔΨm and won’t be affected by other factors such as the oocyte size and the mitochondria number [9]. This study was aimed to explore the influence of vitrification on ΔΨm in human metaphase II (MII) oocytes, and the dynamic changes of ΔΨm after thawing.
Materials and methods
Human oocytes and study design
All oocytes used in this study were donated from IVF patients, that had failed fertilization within 24 h after insemination in Chongqing Reproduction and Genetics Institute from Sep. 2011 to Jan. 2012. All patients had signed consents to agree with the donation of any unfertilized oocytes for research. The study was approved by the Hospital Research and Ethic Board. Age of patients was range from 20 to 35 years (mean ± SD: 30.03 ± 3.16 years), and the cause of infertility was mainly male or tubal factors. Controlled ovarian hyperstimulation protocol was standard long GnRH agonist protocol as previously reported by Ye H et al. [10]. 250ug r-LH (Ovidrel, Serono) were administered 36 h prior to oocytes collection. After retrieval, oocytes were cultured and inseminated in IVF medium. All of the oocytes used in this study were unfertilized MII oocytes in conventional IVF cycles and checked at 24 h after insemination. Those had neither the second polar body nor detectable pronuclei after denuded cumulus and coronal cells by repeated passage though a glass micropipette were defined as failed fertilization oocytes. The failed fertilization ocytes had grossly normal appearance which exhibited a round shape, intact zona pellucida and homogeneous cytoplasm detected under dissecting microscope were involved in this study. The studied oocytes were collected everyday, and then divided into four groups by random number table: non-frozen oocytes as control group (fresh group), three test groups as cultured for 0 h (0 h group), 2 h (2 h group), 4 h (4 h group) after vitrification/thawing. A total of 213 unfertilized MII oocytes from 116 patients were obtained, there were 45, 56, 54, 58 oocytes in fresh, 0 h, 2 h and 4 h group, respectively.
Vitrification/thawing human oocytes
All the vitrification/thawing protocol routinely used in our center were reported previously by Chian RC et al. [11]. The reagent of 1,2-propanediol (PROH), ethylene glycol (EG), sucrose (S) were obtained from Sigma Aldrich; HEPES-buffered human tubal fluid (HEPES-HTF) was obtained from Quinn’s; Serum substitute (SPS), Human serum albumin (HAS), culture medium (G-1), embryo-tested light mineral oil were obtained from Vitrolife.
Vitrification was performed on 3 sequential solutions with increased concentration of cryoprotectants: washing solution (WS), equilibration solution (ES), and vitrification solution (VS). All solutions were prepared in HEPES-HTF supplemented with 20 % SPS which was used as base solution.
Oocytes were placed in WS for 1 min and transferred into ES for 5 min before they were placed in VS for 40 ~ 60 s, and then loaded onto the tip of a vitrification carrier (Cryoloop, HAMPTON) with the minimal VS. The loaded vitrification carrier was plunged directly into liquid nitrogen (LN2) within 30 s. Vitrified oocytes were kept in liquid nitrogen tank for 1 ~ 15 days before thawing.
Thawing was performed in four sequential solutions with decreased concentration of cryoprotectant: thawing solution (TS), diluents solution 1 (DS1), diluents solution 2 (DS2) and holding solution (HS).
TS was balanced at 37 °C before thawing, and other solutions were at room temperature. The vitrified droplet on the carrier’s tip was plunged directly into TS immediately after removing from the LN2. Oocytes were merged into TS droplet for 1 min, then transferred into DS1 for 3 min, DS2 for 5 min and HS for 5 min. Finally, oocytes were placed into embryo culture medium (G-1+ 5%HSA) covered with oil and cultured at 37 °C in humidified atmosphere with 6 % CO2. Oocytes exhibited a rounded shape, homogeneous cytoplasm, intact oolemma and continuous zona pellucida (ZP) were recognized as survival oocytes after thawing.
Oocytes mitochondrial were stained by JC-1
All oocytes in four groups were stained by the ΔΨm-specific probe JC-1 [12]. JC-1 (beyotime, C-2005) was diluted to final concentration of 5 μg/ml in embryo culture medium (G-1 + 5%HSA, Vitrolife). All ooytes were stained in a humidified incubator containing 6 % CO2 at 37 °C for 35 min. The JC-1 reaction were conducted in darkness.
Analysis JC-1 fluorescence in mitochondrial under LSCM
Samples were imaged on a laser scanning confocal microscope (LSCM, Leica, TCS SP2) with fluorescein isothiocyanate (FITC, green) and rhodamine isothiocyanate (RITC, red) channels. Two fluorescent images (one green and one red fluorescence) were acquired in the largest diameter plane of oocytes. Those two images were analyzed by confocal software, which allows for quantitative analysis of the signal intensity of green and red fluorescence. Ratio of RITC to FITC for each oocyte was calculated and used as the endpoint for the ΔΨm [12].
Statistical analysis
The data were presented as mean ± SD of the ratio of red to green fluorescence intensity by ANOVA using SPSS15.0. Differences were considered significant when P < 0.05.
Results
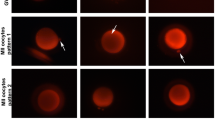
Stained by the mitochondria-specific probes JC-1, MII oocytes can emit red and green fluorescence. Red fluorescence which located around the periphery of oocyte presents higher mitochondrial membrane potential, while green fluorescence that located dispersedly within the oocytes presents lower mitochondrial membrane potential (Fig. 1). The oocytes after vitrification/thawing presented a reduced red fluorescence in periphery, while green fluorescence had no obviously change. These phenomenon are consistent with Jones A et al.’s work [13]. In our study, we found the dispersedly periphery higher mitochondrial membrane potential could gradually recover to the similar level of fresh oocytes after a period of time culture (Fig. 2).

LSCM images of human MII phase oocyte were stained by mitochondria-specific probe JC-1. 1A: a MII oocyte brightfield image. 1B: a MII oocyte stained by JC-I, the red fluorescence was observed in RITC channel. 1C: a MII oocyte stained by JC-I, the green fluorescence was observed in FITC channel

The red fluorescence images on LSCM of human MII oocyte stained by JC-1. 2A, 2B, 2C and 2D represents the typical distribution of red fluorescence in fresh, cultured for 0 h, 2 h and 4 h oocytes after vitrification/thawing, respectively
A total of 168 MII oocytes underwent vitrification/thawing, 75.59 % of them (127/168) survived after thawing. There were 44, 41 and 42 oocytes survived in 0 h, 2 h and 4 h group, respectively. According to Nottola SA et al.’s report [14], good vitrification protocol shouldn’t affect the oocytes shape, ooplasm consistency or vacuole formation. Only survival oocytes which have intact zona pellucida (ZP) and plasmalemma, no overflow or shrinkage of cytoplasm, and no obviously cytoplasm vacuoles (diameter > 5 μm, approximately) were analyzed after stained. According to the above standards, the numbers of oocytes used for the final analysis were 39, 39, 31 and 32 in the fresh group, 0 h, 2 h and 4 h groups, respectively. The mean ratio of red/green fluorescence intensity in the 4 groups was 1.34 ± 0.433, 0.93 ± 0.307, 1.09 ± 0.381 and 1.30 ± 0.467, respectively (Table 1). The ΔΨm of oocytes at 0 h, 2 h groups was statistically lower than the fresh group (P < 0.05), but there was no significant difference between 4 h group and fresh group nor between 0 h group and 2 h group (Fig. 2).
Discussion
In the progress of oocyte maturation, the quantity of mitochondria increases sharply. The MII oocytes contain several hundreds of thousand mitochondria, which provide energy for fertilization and embryo development [15]. Many studies showed that there was a closely association between mitochondria activity and early embryonic development [12, 16–18]. Wilding M et al. reported that the efficiency of mitochondrial respiration in oocytes and preimplantation embryos were correlated with the programmed rate of embryo development [12]. Acton B et al. reported that the differences in mouse embryo competence were related to the ratio of high-to-low polarized mitochondria [16]. ΔΨm is the foundation of mitochondrial respiration. The energy stored in ΔΨm drives the conversion of ADP to ATP by respiratory chain enzymes. The relationship of ΔΨm and oxidative metabolism first described by Mitchell & Moyle (1967) [19], has been studied in somatic cells with ΔΨm -sensitive fluorescent special probes. 5,5′6,6′-tetrachloro-1,1,3,3′-tetraethylbenzimidazolycarbocyanine iodide (JC-1), a particular molecule, has been widely used to assess the relative magnitude of ΔΨm. At relatively lower potential (<100 mv), JC-1 usually exists as a monomer with green fluorescence detected in the fluorescence in isothiocynate (FITC) channel. Whereas, as the potential increases (>140 mv), JC-1 monomers assemble to form metastable stacks or arrays termed J-aggregates with red fluorescence detected in the fluorescein isothiocynate (RITC) channel [9]. Relative ratio of red to green fluorescence intensity was used to represent ΔΨm. Wilding M et al. stained human MII oocytes with JC-1 and detected the intensity of red and green fluorescence. They reported that the ΔΨm value was 1.1 ~ 1.7 [12], which was very similar to our results in this study.
During oocyte cryopreservation/thawing, cryoprotectant toxicity, osmotic stress, non-physiological temperatures, phase transition, pH instability and in-cell ice crystal formation are factors which were believed to affect the structure and function of oocytes. Many studies reported that, cryopreservation can affect subcellular structure of oocytes such as chromosomes, spindle, plasmalemma, cortial granules, mitochondria, and so on [20–22]. ΔΨm is one of the most sensitive indices of freezing injury. It was easily be damaged during the procedure of cryopreservation and thawing. Jones A et al. found that cryopreservation of human MII oocytes were accompanied by loss of the hyperpolarized pericortical mitochondrial domain which is the characteristic of fresh oocyte [13]. Ahn H et al. reported that thawed 2-cell mouse embryos that failed to divide or developed abnormally showed that cryopreservation was associated with loss of mitochondrial hyperpolarization in the subplasmalemmal domain [23]. Most early reports on oocytes cryopreservation used slow frozen protocol. In recent years, vitrification developed rapidly, since it dramatically increased the speed of the freezing and no in-cell ice crystal formed. From these aspects, vitrification probably has the potential to reduce the damage of cryopreservation to oocytes.
EG has low molecular weight, high permeation ability and low toxicity. It has been acknowledged that EG would be the ideal cryoprotectant for oocytes and embryo vitrification [24], and it could retain oocytes developmental competence better than DMSO [25]. Moreover, it has been reported that cryoprotectant mixtures may have some advantage over solutions containing only one permeable cryoprotectant, since PROH and sugar exert major influences on the vitrification properties of ethylene glycol-based solutions and have low toxicity to embryos and oocytes [26]. Because the concentration of vitrification cryoprotectant was higher than that of slow frozen, the equilibration time should be shorter. In this study, we used sucrose, PROH, EG as mixed cryoprotectant for vitrification, 6 mins for equilibration as described previously by Chian RC et al. [11].
After vitrification, we stained human MII oocytes with JC-1 and measured ΔΨm, to observe the change of ΔΨm after thawing and culturing. The result showed that vitrification could temporally reduce the ΔΨm and the periphery red fluoerscence of human MII oocytes in diverse degrees. However, after 4 h incubation followed by thawing, the reduced ΔΨm and the periphery red fluoerscence could spontaneously recover to the similar levels of fresh oocytes. Bianchi V et al. reported that in cryopresered human mature oocytes, the meiotic spindle underwent transient disappearance immediately after thawing, but it could be reorganized in the majority of oocytes after 3–5 h culture [27]. Chen SU et al. reviewed eight published studies mainly in mouse oocytes, and seven of them revealed that oocytes collected immediately after thawing displayed severe disorganization or disappearance of spindles, but recovered to diverse degrees after incubation for 1–3 h at 37 °C[28]. Manipalviratn S et al. evaluated the effect of vitrification and thawing on the intra-oocyte ATP concentration compared to non-vitrified oocytes, and demonstrated that vitrification and thawing procedure has a negative effect on intra-oocyte ATP level, but 3 h incubation after post-thaw, the intra-oocyte ATP level returned to a level significantly higher than that of immediate post-thawed oocytes but still lower than fresh oocytes [29]. From the above studies and our experiment, the recovery time of meiotic spindle reorganization, ATP and ΔΨm are consistent, approximately 3–4 h to recover after thawing. Thus it is possible that fertilization rate may be improved if insemination of oocytes at 4 h after thawing. Chen SU et al. reported that, the fertilization and blastocyst formation rate of vitrified mouse oocytes inseminated at 1 h after thawing was significantly lower than oocytes inseminated at 2 or 3 h after thawing [30]. Recent clinical guidelines suggest that cryopreserved/thawing oocytes need to be cultured for 4 h post thaw before performing ICSI. In the literature, oocytes are inseminated after incubated for at least 1-3 h in most protocols. Porcu E et al. used a slow freezing protocol for oocyte cryopreservation and ICSI was performed in 3 h after thawing. They achieved a fertilization rate of 59 % and a cleavage rate of 91 % [31]. Based on our results and other studies, it is important to culture vitrified/thawed oocytes for 3-4 h to allow spindles, mitochondria and other organelles recovering to normal phase before performing ICSI. Indeed, this study had some limits that we used unfertilized MII oocytes at 24 h after insemination. Thus, more studies particularly using fresh MII oocytes are necessary to verify our finding in the future.
References
Ludwig M, Al-Hasani S, Felberbaum R, Diedrich K. New aspects of cryopreservation of oocytes and embryos in assisted reproduction. Hum Reprod. 1999;14(1):162–85.
Porcu E. Oocyte freezing. Semin Repord Med. 2001;19(3):221–30.
Borini A, Lagalla C, Bonu MA, Bianchi V, Flamigni C, Coticchio G. Cumulative pregnancy rates resulting from the use of fresh and frozen oocytes: 7 years’ experience. Reprod Biomed Online. 2006;12(4):481–6.
Selman H, Angelini A, Barnochi N, Brusco GF, Pacchiarotti A, Aragona C. Ongoing pregnancies after vitrification of human oocytes using a cpmbined solution of ethylene glycol and dimethy sulfoxide. Fertil Steril. 2006;84(4):997–1000.
Cobo A, Diaz C. Clinical application of oocyte vitrification: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2011;96(2):277–85.
Trokoudes KM, Pavlides C, Zhang X. Comparison outcome of fresh and vitrified donor oocytes in an egg-sharing donation program. Fertil Steril. 2011;95(6):1996–2000.
Azambuja R, Petracco A, Okada L, Michelon J, Badalotti F, Badalotti M. Experience of freezing human oocytes using sodium-depleted media. Reprod Biomed Online. 2011;22(1):83–7.
Gualtieri R, Iaccarino M, Mollo V, Prisco M, Iaccarino S, Talevi R. Slow cooling of human oocytes: Ultrastructural injuries and apoptotic status. Fertil Steril. 2009;91(4):1023–34.
Reers M, Smiley S, Mottola-Hartshorn C, Chen A, Lin M, Chen L. Mitochondrial membrane potential monitored by JC-1 dye. In Methods in Enzymology. 1995;260:406–17.
Ye H, Huang GN, Zeng PH, Pei L. IVF/ICSI outcomes between cycles with luteal estradiol (E2) pre-treatment before GnRH antagonist protocol and standard long GnRH agonist protocol: a prospective and randomized study. J Assist Reprod Genet. 2009;26(2–3):105–11.
Chian RC, Kuwayama M, Tan L, Tan J, Kato O, Nagai T. High survival rate of bovine oocytes matured in vitro following vitrification. Reprod Dev. 2004;50(6):685–96.
Wilding M, Dale B, Marino M, di Matteo L, Alviggi C, Pisaturo ML, et al. Mitochondrial aggregation patterns and actvity in human oocytes and preimplantation embryos. Hum Reprod. 2001;16(5):909–17.
Jones A, Blerkom JV, Davis P, Andrew AT. Cryopreservation of metaphase II human oocytes effects mitochondrial membrane potential:implications for developmental competence. Hum Reprod. 2004;19(8):1861–6.
Nottola SA, Coticchio G, Sciajno R, Gambardella A, Maione M, Scaravelli G, et al. Ultrastructural markers of quality in human mature oocytes vitrified using cryoleaf and cryoloop. Reprod Biomed Online. 2009;19 Suppl 3:17–27.
Jansen RP. Germline passage of mitochondria: quantitative considerations and possible embryological sequelae. Hum Reprod. 2000;15 Suppl 2:112–28.
Acton B, Jurisicova A, Jurisica I, Lasper RF. Alterations in mitochondrial membrane potential during preimplantation stages of mouse and human development. Mol Hum Reprod. 2004;10(2):23–32.
Wilding M, Fiorentino A, De Simone ML, Infante V, De Matteo L, Marino M, et al. Energy substrates, mitochondrial membrane potential and human preimplantation embryo division. Reprod BioMed Online. 2002;5(1):39–42.
Wilding M, De Placido G, De Matteo L, Marino M, Alviggi C, Dale B. Chaotic mosaicism in human preimplantation embryos is correlated with a low mitochondrial membrane potential. Fertil Steril. 2003;79(2):340–6.
Mitchell P, Moyle. Chemiosmotic hypothesis of oxidative Phosphorylation. Nature. 1967;213(5072):137–9.
Boiso I, Martí M, Santaló J, Ponsá M, Barri PN, Veiga A. A confocal microscopy analysis of the spindle and chromosome configurations of human oocytes cryopreserved at the germinal vesicle and metaphase II stage. Hum Reprod. 2002;17(7):1885–91.
Coticchio G, Borini A, Distratis V, Maione M, Scaravelli G, Bianchi V, et al. Qualitative and morphometric analysis of the ultrastructure of human oocytes cryopreserved by two alternative slow cooling protocols. Assist Reprod Genet. 2010;27(4):131–40.
Coticchio G, Bonu MA, Sciajno R, Sereni E, Bianchi V, Borini A. Truths and myths of oocyte sensitivity to controlled rate freezing. Reprod Biomed Online. 2007;15(1):24–30.
Ahn H, Sohn I, Kwon H, Jo D, Park Y, Min C. Characteristics of the cell membrane fluidity, actin fibers, and mitochondrial dysfunctions of frozen-thawed two-cell mouse embryos. Mol Reprod Dev. 2002;61(4):466–76.
Shaw JM, Kuleshova LL, MacFarlane DR, Trounson AO. Vitrification properties of solutions of ethylene glycol in saline containing PVP, Ficoll, or dextran. Cryobiology. 1997;35(3):219–29.
Kohaya N, Fujiwara K, Ito J, Kashiwazaki N. High developmental rates of mouse oocytes cryopreserved by an optimized vitrification protocol: the effects of cryoprotectants, calcium and cumulus cells. J Reprod Dev. 2011;57(6):675–80.
Kuleshova LL, MacFarlane DR, Trounson AO, Shaw JM. Sugars exert a major influence on the vitrification properties of ethylene glycol-based solutions and have low toxicity to embryos and oocytes. Cryobiology. 1999;38(2):119–30.
Bianchi V, Coticchio G, Fava L, Flamigni C, Borini A. Meiotic spindle imaging in human oocytes frozen with a slow freezing procedure involving high sucrose concentration. Hum Reprod. 2005;20(4):1078–83.
Chen SU, Lien YR, Chao KH, Ho HN, Yang YS, Lee TY. Effects of cryopreservation on meiotic spindles of oocytes and its dynamics after thawing: clinical implications in oocyte freezing–a review article. Mol Cell Endocrinol. 2003;202(1–2):101–7.
Manipalviratn S, Tong ZB, Stegmann B, Widra E, Carter J, DeCherney A. Effect of vitrification and thawing on human oocyte ATP concentration. Fertil Steril. 2011;95(5):1839–41.
Chen SU, Lien YR, Cheng YY, Chen HF, Ho HN, Yang YS. Vitrification of mouse oocytes using closed pulled straws (CPS) achieves a high survival and preserves good patterns of meiotic spindles, compared with conventional straws, open pulled straws (OPS) and grids. Hum Reprod. 2001;16(11):2350–6.
Porcu E, Fabbri R, Damiano G, Giunchi S, Fratto R, Ciotti PM, et al. Clinical experience and applications of oocyte cryopreservation. Mol Cell Endocrinol. 2000;169(1–2):33–7.
Acknowledgements
We greatly appreciate and thank Dr De-Yi Liu from the Melbourne IVF and the University of Melbourne in Australia, for his comments and revision of the final draft of manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Capsule The article used human unfertilized metaphase II oocytes as material to detect the recovery ability of mitochondrial membrane potential (ΔΨm) after vitrification/thawing. The results showed that the ΔΨm of MII oocytes had temporally dynamic changes within 2h after thawing but could be fully recovered after 4 h culture.
Rights and permissions
About this article
Cite this article
Chen, C., Han, S., Liu, W. et al. Effect of vitrification on mitochondrial membrane potential in human metaphase II oocytes. J Assist Reprod Genet 29, 1045–1050 (2012). https://doi.org/10.1007/s10815-012-9848-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-012-9848-1