
Abstract
Purpose
Stem cell factor (SCF)/c-Kit regulates the proliferation and survival of germ cells or stem cells; however, little is known about the role of SCF/c-Kit in pre-implantation embryo development.
Methods
Using exogenous SCF supplementation and c-Kit siRNA injection, we investigated the role and mechanism of SCF/c-Kit in pre-implantation mouse embryos.
Results
Addition of soluble SCF to the culture medium improved blastocyst formation. c-Kit gene silencing reduced the rate of blastocyst formation and delayed embryonic development. The number of proliferating cells in c-Kit gene-silenced blastocysts decreased, whereas the number of apoptotic cells in blastocysts obtained from both experimental and the control groups was not affected. RT-PCR, immunostaining and western blotting revealed that proliferation-related Akt downstream targets were substantially affected by c-Kit gene silencing.
Conclusion
SCF/c-Kit signaling through Akt downstream targets is likely involved in mediating the cleavage and proliferation of blastomeres during mouse pre-implantation embryogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A number of growth factors including interleukin (IL)-1, IL-6, colony-stimulating factor-1 (CSF-1), tumor necrosis factor-α (TNF-α) and epidermal growth factor (EGF) and their receptors are expressed in pre-implantation embryos and the reproductive tracts of several species [1, 2]. It has been suggested that these factors play important roles in pre-implantation embryonic development and subsequent implantation [3]. Successful implantation requires embryo–uterus interactions that are only initiated when embryonic development is synchronized with the preparation of a receptive uterine state. Although the underlying mechanisms that coordinate the development of blastocysts to implantation-competency with uterine receptivity are not yet fully understood, considerable evidence indicates that soluble growth factors secreted by the uterine epithelium act directly on the embryo to control this process [4]. In turn, developing embryos have been shown to influence the production of a variety of cytokines that may act in an autocrine fashion or may act to modulate endometrial receptivity [5]. The conditions for culturing pre-implantation embryos in vitro are known to differ from the in vivo environment of the female reproductive tract; in particular, in vitro culture systems are deficient in auto/paracrine factors necessary for optimal embryonic development. To overcome these deficiencies, researchers use chemically defined media containing growth factors or developed culture systems employing co-culturing with growth factor-releasing cells [4, 6].
Among the many growth factors, stem cell factor (SCF/Steel factor)/Kit ligand (KL), which acts through the tyrosine kinase receptor, c-Kit, is known for the pleiotropic roles it plays in a variety of cells. It is known that SCF/c-Kit generally signals through the phosphatidylinositol-3-kinase (PI3K)/Akt signal transduction pathway [7, 8]. This pathway mediates a number of cellular processes including regulation of gene transcription, proliferation, differentiation, survival and metabolic homeostasis [7, 9]. Activation of PI3K results in the phosphorylation of the serine-threonine kinase, Akt, which is an important mediator of cell growth, survival and migration [10].
Just prior to endometrial attachment, c-Kit is expressed from the late two-cell stage to the expanded and hatched blastocyst stage [1]. Furthermore, c-Kit is detected in the blastocyst and outgrowth blastocysts as well as in endometrial cells [11]. This finding is also confirmed through immunohistochemical studies of the trophectoderm and the inner cell mass of mouse blastocysts. During the pre-implantation period, SCF transcripts are detected in the uterus, suggesting that these growth factors may act through a paracrine mechanism. In fact, recombinant SCF added to culture medium acts through c-Kit to enhance mouse embryonic development and reduces the detrimental effects of factor deficiencies in in vitro culture systems [6, 12, 13]. Nevertheless, our understanding of the specific roles of SCF/c-Kit signaling in pre-implantation embryos remains superficial due to the difficulty of analyzing mammalian embryos.
RNA interference (RNAi) is an established powerful experimental tool for analyzing gene function in mammalian cells and embryos [14–17]. Applying a similar approach, we used an siRNA strategy to explore the specific function of the SCF/c-Kit system in mouse pre-implantation embryonic development.
Materials and methods
Embryo collection
ICR female (4–6-week-old) mice used for this study were housed in a temperature- and humidity-controlled room under a 12 h/12 h light/dark cycle, and food and water were provided ad libitum. Protocols for the use of animals in these experiments were approved by the Institutional Animal Care and Use Committee of Cha University (IACUC080012). Female ICR mice were superovulated by injection of 5 IU pregnant mare serum gonadotropin (PMSG; Folligon, Intervet Co., Holland) followed after 48 h by an injection of human chorionic gonadotropin (hCG; Chorulon, Intervet Co., Holland). Next, the mice were mated overnight with 8–10-week-old males of the same strain. At 20–22 h post-hCG, we collected one-cell embryos from excised oviducts into modified human tubal fluid medium (HTF; Irvine Scientific, Santa Ana, CA). Mouse embryos were pooled and randomly distributed into the different experimental groups.
Embryo culture
Cooperative interactions of SCF in mouse embryonic development were studied by culturing five to ten one-cell embryos in 25 μl of modified serum-free KSOM medium with or without SCF (100 ng/ml; R&D Systems, Inc.). Additionally, cultured embryos in KSOM medium containing 3 mg/ml of BSA was used as a positive control. All embryos of each group were cultured for 72 h (96 h post-hCG) in a humidified 5% CO2 atmosphere at 37°C. The rate of embryonic development compared to the hatching stage was monitored at 96 h post-hCG.
Microinjection of siRNA
Chemically synthesized commercially obtained 21-nucleotide siRNAs (Bioneer, Seoul, Korea) were selected based on published nucleotide sequences (GenBank Accession No. NM_021099 for c-Kit) and were designed to form 19-bp siRNA with 2-deoxynucleotide overhangs at both 3′-ends (CGA CCT TTT ATA GGC ACG T).
siRNAs were microinjected into the cytoplasm of one-cell embryos using a constant-flow system (Transjector; Eppendorf, Hamburg, Germany), which delivers each 10 pl siRNA (final concentration 100 pmol) via an injection pipette loaded with siRNA solution. Sham-injected control embryos were microinjected with vehicle (siRNA duplex buffer, RNAse free, pH 7.4; Bioneer, Seoul, Korea). After microinjection, the embryos were cultured in serum-free KSOM medium containing 100 ng/ml SCF.
Isolation of total RNA and real-time RT-PCR
Embryos were washed three times with Ca2+-/Mg2+-free Dulbecco’s phosphate-buffered solution (DPBS; Gibco BRL, Grand Island, NY) and treated with distilled water treated with 0.1% diethyl-pyrocarbonate (DEPC; Sigma-Aldrich). Total RNA from 30 washed blastocysts (96 h post-hCG) was extracted for real-time RT-PCR using TRIzol (Gibco BRL) as described in the manufacturer’s instructions.
Total RNA from 30 blastocysts (in 10 μl) was incubated for 30 min at 37°C with 5 mmol/l MgCl2 and 1 U of DNase I. Next, the RNA was reverse transcribed by adding 1 mmol/l dNTP, 2.5 μmol/l oligo-dT and 2.5 U reverse transcriptase (Superscript; Invitrogen), and the mixture was incubated in a Mastercycler Gradient (Eppendorf) at 42°C for 60 min and then at 99°C for 5 min. After the reaction was completed, cDNA was either used directly for real-time PCR or stored at −20°C. The following mRNA targets were amplified (with the indicated PCR primer pairs): c-Kit (forward primer, 5′-CTG GTG GTT CAG AGT TCC ATA GAC-3′; reverse primer, 5′- TCA ACG ACC TTC CCG AAG GCA CCA -3′); mTOR (mammalian target of rapamycin; forward primer, 5′-CTT TGG CCT GGT GAA CAC AC-3′; reverse primer, 5′-CAT CAC AGT GTG GCA TGT GG-3′), tuberin (forward primer, 5′-TTC CTA CAG CTC TAC CAT TCG C-3′; reverse primer, 5′-TTA ATG GTG CCC AGT TTG AAG T-3′), IKK (IκBα kinase; forward primer, 5′-TGA TGC TTA TGT GGC ACC CT-3′; reverse primer, 5′-GGG CTC CTG AAG GAT ACA GC-3′), and Bad (forward primer, 5′-CCA GAG TTT GAG CCG AGT GA-3′; reverse primer, 5′-CAC CAG GAC TGG ATA ATG CG-3′). The levels of target mRNA were quantified relative to the levels of mRNA for the housekeeping 18S ribosomal protein (forward primer, 5′-AGA TGA TCG AGC CGC GC-3′; reverse primer, 5′-GCT ACC AGG GCC TTT GAG ATG GA-3′). In preliminary studies, 18S mRNA levels in embryos were unaffected by the culture conditions. Amplification products were quantified on a DNA Engine 2 fluorescence detection system (MJ research) using the DyNAmo SYBR green qPCR kit (Finnzymes, Espoo, Finland). Reactions were performed in a reaction mix containing 4 μl DEPC-treated water, 2 μl forward primer (5 pmol), 2 μl reverse primer (2 pmol), 10 μl premix with SYBR Green, and 2 μl cDNA template in a total volume of 20 μl. The PCR protocol used an initial denaturing step of 95°C for 10 min followed by 45 cycles of denaturation at 95°C for 30 s, annealing of primers at 60°C (18S), 58°C (c-Kit), 60°C (mTOR), 60°C (tuberin), 55°C (IKK) or 60°C (Bad) for 30 s, and extension at 72°C for 30 s. Fluorescence was measured at the end of each cycle during the 72°C step. In the final step, a melting curve was generated by raising the temperature from 65°C to 95°C at a rate of 0.1°C/s, with constant measurement of fluorescence, followed by cooling at 40°C for 30 s. Relative gene expression was quantified using the 2-∆∆CT method.
Embryo fixation and immunostaining
To investigate c-Kit, Akt and phosphorylated-Akt (p-Akt) localization and expression levels in mouse embryos, we performed immunostaining using an anti-c-Kit monoclonal antibody (Santa Cruz Biotechnology, CA, USA), anti-Akt monoclonal antibody (R&D System, CA, USA) and anti-phosphorylated-Akt polyclonal antibody (Santa Cruz Biotechnology, CA, USA). Sham- and c-Kit siRNA-microinjected embryos were washed three times in DPBS with polyvinylpyrrolidone (PVP, 1 mg/ml), and five to ten embryos per stage were fixed in a 20-μl drop of paraformaldehyde (1% v/v in DPBS) for 1 h. Embryos were incubated in 0.1% Triton X-100 in DPBS for 1 h to permeabilize the samples. Next, the samples were washed three times with DPBS and then incubated in blocking solution (1% BSA in DPBS) for 30 min at room temperature to suppress nonspecific antibody binding. After washing three times with DPBS, the fixed samples were incubated with each antibody diluted to 1:200 (c-Kit: 2 μg/ml, p-Akt: 2 μg/ml and Akt: 1 μg/ml, independently) with DPBS containing 0.1% Tween-20 and 3% BSA for 60 min at room temperature or overnight at 4°C. Next, the samples were incubated with a rhodamine-conjugated anti-rabbit IgG antibody (KPL) and fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG antibody (KPL) diluted to 1:500 with DPBS for 60 min at room temperature. Finally, embryos were counterstained with 1 μg/ml 4′,6′-diamidino 2-phenyindiol (DAPI; Sigma-Aldrich). Following multiple washes, dot slides were mounted in Vectashield mounting medium (Vector laboratories, Burlingame CA). Stained samples were viewed on an inverted confocal laser scanning fluorescence microscope (LSM 510; Carl Zeiss, Oberkochen, Germany) at 400× magnification. Micrographs were stored in the Zeiss LSM Image Browser version 2.30.011 (Carl Zeiss Jena GmbH, Jena, Germany).
Detection of proliferation and apoptosis
The proliferation of embryonic cells was assessed by staining for proliferating cell nuclear antigen (PCNA). Fixed embryos were washed, transferred to a solution containing monoclonal anti-PCNA antibody (1 μg/ml, clone PC-10; DAKO, Glostrup, Denmark), and incubated for 1 h at room temperature. Embryos were then incubated with FITC-conjugated secondary antibody for 1 h and then with 1 μg/ml DAPI in DPBS for 1 h.
The embryos were washed twice with 1% BSA in DPBS and mounted onto glass slides using Vectashield. The terminal dUTP nick-end labeling (TUNEL) assay was used to assess the presence of apoptotic cells (in situ Cell Death Detection Kit, TMR red; Roche Diagnostics, Indianapolis, IN). After washing three times with DPBS, blastocysts were fixed with 1% paraformaldehyde solution in DPBS for 1 h and permeabilized with 0.2% Tween-20 in DPBS for 1 h. Fixed embryos were incubated in TUNEL reaction medium (50 μl) for 1 h at 37°C in the dark. After the reaction was stopped, total cell numbers in blastocysts were determined by counterstaining with DAPI. Mounted dot slides were stored at −20°C for up to 7 days before qualitative fluorescence microscopic examination and subsequent confocal laser scanning microscopic analysis. Reconstructed confocal images were used to measure the number of red-spot containing nuclei (apoptotic cells) per total cells (blue nuclei) in each embryo.
Western blot analysis
To compare p-Akt expression in blastocysts of sham-operated or c-Kit gene-silenced groups, proteins were extracted from 50 pooled blastocysts for western blotting. Extract samples were separated on SDS-PAGE and transferred to nitrocellulose membranes (Amersham Biosciences, Little Chalfont, UK). Nonspecific binding sites were blocked by incubating membranes overnight at 4°C in blocking buffer containing 5% skim milk. Membranes were washed three times in TBST and incubated for 1 h at 24°C with a 1:200 (2 μg/ml) dilution of rabbit polyclonal anti-phospho-Akt antibody (Santa Cruz Biotechnology, CA, USA) and anti-c-Kit monoclonal antibody (Santa Cruz Biotechnology, CA, USA) in blocking buffer. Membranes were washed three times in TBST (0.3% Tween-20) and incubated with peroxidase-conjugated anti-rabbit IgG (1:200) for 60 min at 24°C in blocking buffer. Membranes were washed three times in TBST, and protein bands were detected using the enhanced chemiluminescence detection method (ECL; Amersham, Inc.).
Statistics
Unless otherwise specified, each experiment was carried out using at least three replicates. The data are expressed as the mean±SEM. Statistical significance of differences among treated groups were evaluated by one-way analysis of variance (ANOVA) using a log-linear model in the Statistical Analysis System (SAS, Cary, NC, USA). Values of P < 0.05 were considered statistically significant.
Results
Effects of in vitro supplementation with SCF
To elucidate the effects of SCF on the development of pre-implantation mouse embryos, we cultured one-cell embryos in the presence or absence of SCF or 3 mg/ml BSA. As shown in Supplemental Figure 1, the addition of 100 ng/ml SCF increased the rate of embryo development compared to the absence of SCF. Moreover, the effect of 100 ng/ml SCF was significantly greater than the effect of 1 or 10 ng/ml SCF (data not shown). SCF supplementation (100 ng/ml) increased the blastocyst formation rate within a given period of time at 96 h post-hCG (47.74 ± 8.7% vs. 64.09 ± 9.8%) (Supplemental Fig. 1). Specifically, blastocyst and expanded blastocyst rates were significantly higher in groups supplemented with SCF (21.53 ± 1.2% and 21.73 ± 4.3%) compared to the group not treated with SCF (9.59 ± 1.1% and 11.19 ± 3.1%, p < 0.05).
The relationship between embryonic development and siRNA-mediated downregulation of c-Kit protein expression
To test the effects of c-Kit siRNA on pre-implantation stage embryonic development, we microinjected the cytoplasm of one-cell embryos with c-Kit siRNA (see “Materials and Methods”) and then evaluated the embryos after culturing in serum-free KSOM medium containing 100 ng/ml SCF for an additional 72 h (96 h post hCG). As shown in Fig. 1, the injection of c-Kit siRNA effectively inhibited pre-implantation development. Specifically, the frequency of blastocyst and hatching blastocyst stages were reduced compared to sham-injected groups (33.16 ± 1.0% vs. 15.67 ± 2.6% and 11.00 ± 3.1% vs. 3.00 ± 1.0%, p < 0.05, respectively).

Effect of siRNA-mediated c-Kit downregulation on the development of mouse one-cell embryos. After microinjection, the embryos were cultured in serum-free KSOM medium containing 100 ng/ml SCF. The data are presented as the mean±SEM. a,b Values within the same column with different superscripts are significantly different (P < 0.05). Note: Control (116 zygotes, uninjected group), Sham (112 zygotes, vehicle-injected group), c-Kit siRNA (161 zygotes, CGA CCT TTT ATA GGC ACG T target siRNA-injected group). Early Bla: early blastocyst (less than 50% cavity), Bla: blastocyst (greater than 50% cavity), Expanded Bla: expanded blastocyst, Hatching Bla: hatching blastocyst
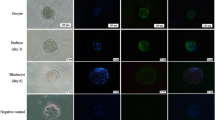
Using RT-PCR, we found that c-Kit mRNA was strongly expressed in the sham-injected group but was reduced in the c-Kit siRNA-injected groups (Fig. 2a). Real-time RT-PCR showed that c-Kit siRNA-injected groups significantly reduced c-Kit mRNA expression than sham-injected group (fold change 1.00 ± 0.1 vs 3.59 ± 0.87, p < 0.05, respectively) (Fig. 2b). Additionally, Western blotting results showed that c-Kit protein was strongly expressed in the sham-injected group but was reduced in the c-Kit siRNA-injected groups (Fig. 2c). To confirm if the inhibitory effects of c-Kit siRNA on embryonic development were related to the silencing of the c-Kit gene, we evaluated c-Kit protein expression levels in mouse embryos at each stage of development (i.e., two-cell, morula and blastocyst) by immunostaining. In two-cell embryos treated with c-Kit siRNA, fluorescence intensity was similar to the level in the sham-injected group; however, from morula to blastocysts, fluorescence signals in the embryos of the c-Kit siRNA group were much weaker than the level in the sham-injected embryos (Fig. 2d).

Specific reduction of c-Kit mRNA and protein expression by siRNA injection. Quantification of c-Kit mRNA in mouse blastocysts by RT-PCR (a) and real-time RT-PCR (b). c-Kit siRNA or sham (vehicle) was injected into one-cell mouse embryos. Injected and control embryos were cultured in serum-free KSOM medium containing 100 ng/ml SCF. Total RNA isolated from blastocyst stage embryos was reverse transcribed and amplified. c Western blot of c-Kit in extracts from 50 blastocysts in the sham- and c-Kit siRNA-injected groups. d Embryos at each stage were fixed and stained with an anti-c-Kit antibody. Right, lower panel depicts a blastocyst stained with secondary antibody only as a negative control. Sham and c-Kit siRNA groups were analyzed at the same exposure time and more than 10 embryos were used for the fluorescence quantitation
Effect of siRNA-mediated c-Kit inhibition on blastomere proliferation and survival
To determine if the inhibition of c-Kit affected the proliferation and apoptosis of blastomeres, we used PCNA staining and TUNEL assays to determine the number of proliferating and apoptotic cells in blastocysts derived from c-Kit siRNA- and sham-injected embryos at 96 h post-hCG. A significant decrease in the percentage of proliferating cells was found in the c-Kit siRNA group (62.7 ± 7.0) at the end of the culture period compared to the percentage in the sham-injected group (73.3 ± 6.2, p < 0.05). The percentage of apoptotic cells per blastocyst was not different between the two groups (5.1 ± 3.5 and 4.0 ± 2.5, p > 0.05) (Fig. 3).

Effect of c-Kit siRNA on proliferation and apoptosis of blastomeres. a Proliferating blastomeres were detected by an anti-PCNA antibody (green). b Apoptosis of blastomeres was detected by TUNEL assay (TMR-red staining, arrows indicate apoptotic cells). Graphical representation of the results in panels A and B show the percentage of proliferating cells (c; PCNA) and the percentage of apoptotic cells (c; TUNEL) that developed into blastocysts following injection of c-Kit siRNA. The data are presented as the mean±SEM. a,b Values within the same column with different superscripts are significantly different (P < 0.05). Sham and c-Kit siRNA groups were analyzed at the same exposure time and more than 10 embryos (Sham; n = 33 and c-Kit siRNA: n = 33) were used for the cell number count
Effect of inhibition of c-Kit expression on Akt phosphorylation
To compare the effect of c-Kit inhibition on the Akt pathway, we determined the p-Akt protein expression levels in mouse embryos at the blastocyst stage (96 h post-hCG) using immunostaining and western blotting in two groups (the sham-injected group and the c-Kit siRNA groups). In the blastocysts, total Akt fluorescence signals were not different between the two groups; however, the p-Akt protein fluorescence intensities were higher in the sham-injected group compared to the c-Kit siRNA groups (Fig. 4a). Western blotting produced similar results that demonstrated that the relative detection of β-actin was not different; however, the p-Akt bands were much weaker in the c-Kit siRNA groups compared to the sham-injected group (Fig. 4b).

a Specific reduction of total Akt and p-Akt protein expression by c-Kit siRNA. Blastocyst stage embryos were fixed and stained with an anti-p-Akt and anti-total Akt antibody. Changes in total Akt and p-Akt protein levels were evaluated following injection of sham and c-Kit siRNA into one-cell mouse embryos. Sham and siRNA groups at the same exposure length. b Western blot of p-Akt in extracts from 50 blastocysts in the sham- and c-Kit siRNA-injected groups. c PCR band shows semi-quantitative RT-PCR results performed prior to real-time RT-PCR (Negative: no reverse transcriptase). d Quantification of mRNA for the downstream targets of Akt (mTOR, tuberin, IKK and Bad) in mouse blastocysts by real-time RT-PCR. The data are presented as the mean±SEM. a,b Values within the same column with different superscripts are significantly different (P < 0.05)
Real-time RT-PCR analysis of Akt downstream genes in embryos injected with c-Kit siRNA
To understand the mechanism by which SCF/c-Kit signaling mediates its effects on mouse pre-implantation embryogenesis, we quantified the mRNA expression of Akt downstream targets such as mTOR, tuberin, IKK and Bad in one-cell embryos microinjected with c-Kit siRNA. If the expression of c-Kit is suppressed by the siRNA, the expression levels of these downstream genes should decrease or increase. In blastocyst stage embryos, using real-time RT-PCR, we found that c-Kit siRNA injection downregulated the mRNA expression of the cell proliferation-related Akt target, mTOR, and increased the expression of tuberin mRNA, which is an Akt target (mTOR) inhibitor (p < 0.05). The mRNA of the cell apoptosis-related Akt targets, IKK and Bad, were not significantly different between the two groups (Fig. 4d). Furthermore, these results were reconfirmed by RT-PCR (Fig. 4c).
Discussion
SCF and its cognate receptor, c-Kit, are known to have important roles in mammalian reproduction. The essential role of SCF/c-Kit in oocyte growth and follicular development has been a topic of investigation for decades [18, 19]. c-Kit is expressed at the surface of mammalian oocytes at all stages of follicular development in postnatal ovaries of mice, rats, and humans [20, 21], whereas SCF/KL is produced only by granulosa cells [7, 22]. In fact, treatment of in vitro cultured follicles from 8-day-old mice with SCF/KL significantly increased the growth rate of oocytes [23]. Additionally, an anti-c-Kit antibody (ACK2) that interferes with the binding of SCF/KL to c-Kit was found to severely inhibit the growth of late fetal and neonatal oocytes co-cultured with ovarian cells. In addition, this c-Kit neutralizing antibody disturbs the onset of primordial follicle development, primary follicle growth, follicular fluid formation of preantral follicles, and penultimate-stage ovarian follicle maturation before ovulation in vivo, indicating a sequential requirement for Kit in ovarian follicle development in mice [24].
The level of c-Kit mRNA increases following fertilization, and the mRNA is continuously expressed throughout pre-implantation development [1]. SCF/c-Kit-mediated signaling cascades in mammalian pre-implantation embryos are still poorly understood. Generally, activated, tyrosine-phosphorylated Kit recruits PI3K from the cytoplasm to the cell membrane region via binding of its phospho-tyrosine moiety to the Src homology 2 (SH2) domain of PI3K, which activates PI3K [25, 26]. A well-known downstream target of PI3K is the serine/threonine kinase, Akt, which transmits survival and proliferation signals from growth factors [27, 28]. Numerous Akt substrates have been identified and validated [29]. Activated Akt phosphorylates target molecules that control key cellular processes such as apoptosis, cell cycle progression, transcription and translation. The best-studied downstream substrate of Akt is the serine/threonine kinase, mTOR. Akt can directly phosphorylate and activate mTOR, which modulates cell proliferation, in part, by regulating initiation of mTOR translation. Additionally, Akt indirectly activates mTOR by phosphorylating and inactivating tuberin, which normally inhibits mTOR through the GTP-binding protein, Rheb (Ras homolog enriched in brain). Additionally, Akt phosphorylates and inactivates the pro-apoptotic protein, Bad, which controls release of cytochrome c from mitochondria and regulates apoptosis [30]. In contrast, Akt can phosphorylate IKK, which indirectly increases the activity of nuclear factor kappa B (NF-κB) and stimulates the transcription of pro-survival genes [31, 32]. We inferred that SCF/c-Kit signaling in pre-implantation embryos may regulate certain common pathways that are involved in cell proliferation and apoptosis. Accordingly, in the present study, we sought to determine which downstream genes of SCF/c-Kit were affected after knock-down of the c-Kit gene in mouse pre-implantation embryos, focusing specifically on Akt, mTOR, tuberin, IKK and Bad. After microinjecting c-Kit siRNA, mRNA levels of the cell proliferation-associated genes, mTOR and tuberin, were significantly changed in the blastocyst. In contrast, the expression of IKK and Bad, genes associated with apoptosis, was not different from control embryos (Fig. 4). PCNA immunostaining also showed that the percentage of proliferating cells decreased in siRNA-microinjected embryos (Fig. 3). Moreover, we observed that the addition of soluble SCF into the culture medium improved the development of pre-implantation mouse embryos, resulting in accelerated blastocyst formation and hatching (Fig. S1). These results may suggest that SCF/c-Kit function is involved in the proliferation of pre-implantation mouse embryos. Although SCF has been shown to exert an anti-apoptotic effect on poorly differentiated cells such as hematopoietic progenitor cells or germinal progenitor cells [21], this anti-apoptotic action does not appear to be involved in the pre-implantation embryo. The absence of a difference in the number of apoptotic cells between blastocysts derived from c-Kit siRNA-injected and control embryos confirms this finding. Collectively, these data support the interpretation that SCF/c-Kit signaling primarily contributes to proliferation speed in the pre-implantation mouse embryo.
The effect of SCF supplementation has been well studied by a number of researchers. During the co-culturing of murine embryos with a human ovarian granulosa tumor-derived cell line (KGN cells), it was observed that adding human recombinant SCF to the medium significantly enhanced embryo development to the late blastocyst and hatching stages [6]. In a separate study, the growth factors (IGF1, IGF2, SCF and EGF) reduced the number of apoptotic cells and increased the rate of development in embryos exposed to hydrogen peroxide [33]. The results presented in the current study suggest that adding SCF to the medium may compensate for the depletion of secreted factors produced in vivo in the reproductive tract, reversing the delay of in vitro development of pre-implantation embryos and resulting in higher hatching rates (Fig. S1). Further study of the involvement of a potential paracrine mechanism in the release of this growth factor from the reproductive tract may contribute to the development of chemically defined media for pre-implantation embryos.
In conclusion, SCF/c-Kit signaling may be involved in mediating the proliferation of blastomeres in pre-implantation mouse embryos via a mechanism involving Akt downstream targets. Our results also suggest that the SCF/c-Kit-PI3K-Akt-tuberin-mTOR cascade may make an important contribution to the regulation of the in vitro development of mouse pre-implantation embryos.
References
Arceci RJ, Pampfer S, Pollard JW. Expression of CSF-1/c-fms and SF/c-kit mRNA during preimplantation mouse development. Dev Biol. 1992;151:1–8.
Paria BC, Dey SK. Preimplantation embryo development in vitro: cooperative interactions among embryos and role of growth factors. Proc Natl Acad Sci USA. 1990;87:4756–60.
Rappolee DA, Brenner CA, Schultz R, Mark D, Werb Z. Developmental expression of PDGF, TGF-alpha, and TGF-beta genes in preimplantation mouse embryos. Science. 1988;241:1823–5.
Tartakovsky B, Ben-Yair E. Cytokines modulate preimplantation development and pregnancy. Dev Biol. 1991;146:345–52.
Sharkey AM, Dellow K, Blayney M, Macnamee M, Charnock-Jones S, Smith SK. Stage-specific expression of cytokine and receptor messenger ribonucleic acids in human preimplantation embryos. Biol Reprod. 1995;53:974–81.
Taniguchi F, Harada T, Nara M, Deura I, Mitsunari M, Terakawa N. Coculture with a human granulosa cell line enhanced the development of murine preimplantation embryos via SCF/c-kit system. J Assist Reprod Genet. 2004;21:223–8.
Linnekin D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31:1053–74.
Ronnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004;61:2535–48.
van Dijk TB, van Den Akker E, Amelsvoort MP, Mano H, Lowenberg B, von Lindern M. Stem cell factor induces phosphatidylinositol 3′-kinase-dependent Lyn/Tec/Dok-1 complex formation in hematopoietic cells. Blood. 2000;96:3406–13.
Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305.
Mitsunari M, Harada T, Tanikawa M, Iwabe T, Taniguchi F, Terakawa N. The potential role of stem cell factor and its receptor c-kit in the mouse blastocyst implantation. Mol Hum Reprod. 1999;5:874–9.
Glabowski W. The protective effect of stem cell factor (SCF) on in vitro development of preimplantation mouse embryos. Ann Acad Med Stetin. 2005;51:83–93.
Glabowski W, Kurzawa R, Wiszniewska B, Baczkowski T, Marchlewicz M, Brelik P. Growth factors effects on preimplantation development of mouse embryos exposed to tumor necrosis factor alpha. Reprod Biol. 2005;5:83–99.
Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science (New York, NY). 1999;286:950–2.
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8.
Bernstein E, Denli AM, Hannon GJ. The rest is silence. Rna. 2001;7:1509–21.
Khang I, Sonn S, Park JH, Rhee K, Park D, Kim K. Expression of epithin in mouse preimplantation development: its functional role in compaction. Dev Biol. 2005;281:134–44.
Besmer P, Manova K, Duttlinger R, Huang EJ, Packer A, Gyssler C, et al. Development (Cambridge, England). 1993;125:37.
Eppig JJ. Oocyte control of ovarian follicular development and function in mammals. Reproduction. 2001;122:829–38.
Horie K, Takakura K, Taii S, Narimoto K, Noda Y, Nishikawa S, et al. The expression of c-kit protein during oogenesis and early embryonic development. Biol Reprod. 1991;45:547–52.
Driancourt MA, Reynaud K, Cortvrindt R, Smitz J. Roles of KIT and KIT LIGAND in ovarian function. Rev Reprod. 2000;5:143–52.
Ismail RS, Okawara Y, Fryer JN, Vanderhyden BC. Hormonal regulation of the ligand for c-kit in the rat ovary and its effects on spontaneous oocyte meiotic maturation. Mol Reprod Dev. 1996;43:458–69.
Packer AI, Hsu YC, Besmer P, Bachvarova RF. The ligand of the c-kit receptor promotes oocyte growth. Dev Biol. 1994;161:194–205.
Yoshida H, Takakura N, Kataoka H, Kunisada T, Okamura H, Nishikawa SI. Stepwise requirement of c-kit tyrosine kinase in mouse ovarian follicle development. Dev Biol. 1997;184:122–37.
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7.
Stokoe D. The phosphoinositide 3-kinase pathway and cancer. Expert Rev Mol Med. 2005;7:1–22.
Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014.
Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–29.
Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–41.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41.
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–5.
Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ. Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol. 1999;1:500–6.
Kurzawa R, Glabowski W, Baczkowski T, Wiszniewska B, Marchlewicz M. Growth factors protect in vitro cultured embryos from the consequences of oxidative stress. Zygote. 2004;12:231–40.
Acknowledgements
This research was supported by a grant (2009-0093821) from Priority Research Centers Program funded by the Ministry of Education, Science and Technology, Republic of Korea.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Capsule
c-Kit siRNA inhibited development of preimplantation-stage mouse embryo and changed the mRNA expression of Akt target gene (upregulation of Tuberin and downregulation of mTOR). Tuberin normally inhibited mTOR which modulates cell proliferlation.
Jung Jin Lim and Jin Hee Eum contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Figure 1
Effect of SCF supplementation on the development of mouse one-cell embryos at 96 h post-hCG. The data are expressed as the mean±SEM. a,b Values within the same column with different superscripts are significantly different (P < 0.05). Note: KSOM (only): 139 zygotes, serum-free KSOM media, KSOM (100 ng/ml SCF): 138 zygotes, serum-free KSOM media with 100 ng/ml soluble SCF, KSOM (3 mg/ml BSA): 144 zygotes, KSOM media with 3 mg/ml BSA. Early Bla: early blastocyst (less than 50% cavity), Bla: blastocyst (greater than 50% cavity), Expanded Bla: expanded blastocyst, Hatching Bla: hatching blastocyst. (GIF 170 kb)
Rights and permissions
About this article
Cite this article
Lim, J.J., Eum, J.H., Lee, J.E. et al. Stem cell factor/c-Kit signaling in in vitro cultures supports early mouse embryonic development by accelerating proliferation via a mechanism involving Akt-downstream genes. J Assist Reprod Genet 27, 619–627 (2010). https://doi.org/10.1007/s10815-010-9449-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-010-9449-9