
Abstract
A rapid detection technology for okadaic acid (OA) in shellfish with one-step immunochromatographic assay using colloidal gold-labeled monoclonal antibody (Mab) probe was developed. OA is one of the diarrhetic shellfish toxins. Firstly, OA was conjugated to bovine serum albumin, and the conjugations as immunogen were injected into mice to raise the polyclonal antibody against OA. Hybridoma cells fused between spleen cells from immunized mouse and myeloma cells (Sp2/0) were prepared and injected into mice intraperitoneally at 1 × 106 cells to produce monoclonal antibody in the ascitic fluid. With the monoclonal antibody against OA, the idc-ELISA assay was established to detect OA. The calibration curve for OA was linear over the concentration range of 0.31–50 ng mL−1, and the detection limit for OA was 0.45 ng mL−1. On that basis, paper test strips for detecting OA were prepared, and a fast detection method for okadaic acid using gold-labeled immunological assay was established. With the paper test strips, the detection limit was 6.25 ng mL−1, and whole detection process for OA in shellfish samples needed only about 40 min.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Okadaic acid (OA) is a lipophilic marine algal toxin, which was first extracted from the sponges Halichondria okadai and Halichondria melanodocia (Tachibana et al. 1981). Later it was found to be the same toxin as that isolated from the dinoflagellates Prorocentrum and Dinophysis (Murakami et al. 1982; Carmody et al. 1996). OA is one of the major toxins responsible for diarrhetic shellfish poisoning (DSP) (Murata et al. 1982) and can be accumulated in shellfish feeding on toxigenic algae cells. Diarrhetic shellfish poisoning is the term used to describe the rapid onset of gastrointestinal symptoms such as vomiting and diarrhea in people who consume toxic shellfish (Amzil et al. 1992; Souto et al. 2001). In addition, OA potentially can facilitate tumor promotion. However, the medical treatment for aforementioned symptoms has not been confirmed. Treatment with strong mineral acids could destroy OA in the shellfish tissue, but the toxin compounds are rather stable during normal cooking procedures (Garthwaite 2000).
Detection techniques of OA have been classified into three categories: biological methods, physical chemical methods, and biochemical methods. Biological methods (e.g., mouse bioassays) have been almost completely replaced by alternative methods due to insufficient accuracy or humanism (Mouratidou et al. 2006). Physical chemical methods such as liquid chromatography–mass spectrometry (LC–MS)/MS are commonly used to determine OA and related derivatives simultaneously with multiple reaction monitoring instead of selected ion monitoring. LC-MS/MS is particularly useful to reveal important information on the chemical structure of new toxins (Quilliam 2003) in contaminated shellfish tissue. However, the entire analysis procedure requires a long time, complicated pretreatment steps and trained personnel. Moreover, instrumentation required for these methods is not suitable for on-site measurement. Therefore, there is a growing demand for more rapid, reliable, and economical methods for the qualification and quantification of OA residues.
Biochemical techniques including immunoassays are simpler to perform than the complicated physical chemical methods. Enzyme-linked immunosorbent assay (ELISA) shows a high sensitivity and excellent specificity. Hybridization techniques are used to generate MAbs against OA with high sensitivity. However, they have some cross-reactivity against other DTX analogs (Morton and Tindall 1996). Therefore, the specificity of the monoclonal antibody is crucial for assay reliability; MAbs with excessive specificity do not recognize other members of one toxin family and can underestimate the total toxin content; however, low specificity would lead to detection of non-toxic analogs. Commercial ELISA test kits such as UBE ELISA (Ube Industries, Japan), Rougier ELISA (Rougier Bio-Tech, Canada), and DSP-Check, (R-Biopharm, Germany) are used to analyze samples containing OA and no DTX1 in some countries (Morton and Tindall 1996; Mouratidou et al. 2006). As a potential complementary strategy, an electrochemical immunosensor based on MAbs has been developed for the OA detection in the analysis of contaminated shellfish samples, providing some advantages over other immunoassays in terms of analysis time, selectivity, sensitivity, and cost (Mònica Campàs et al. 2008).
Nowadays the colloidal gold immunochromatography assay is widely used in many fields, such as hospitals, inspection departments, and local medical offices. Colloidal gold-labeled antigen or antibody is used as the tracer. This system was devised in order to determine the concentration of the measured subjects conveniently and rapidly. It is easy and cheap to prepare in the laboratory. The color of colloidal gold is intense, and no development process is required for visualization. The label is very stable in liquid or dried form and does not bleach after staining on nitrocellulose membrane. Moreover, this improved method provides a reference to the development of colloidal gold immunochromatography strip for detecting other pesticide and poison chemicals. Nonetheless, the application of the colloidal gold immunochromatography assay method for OA in shellfish samples has not been reported so far. Here we developed an indirect competitive enzyme-linked immunosorbent assay (idc-ELISA) and a colloidal gold immunochromatography test strip method for OA fast detection by preparing a monoclonal antibody against OA.
Materials and methods
Certified OA, dinophysistoxin DTX-1, saxitoxin (STX), and microcystin-LR (MC-LR) were purchased from Express Technology Co Ltd. (Taiwang, China). Horseradish peroxidase (HRP)-labeled goat anti-mouse IgG antibodies and 3,3′,5,5′-tetramethylbenzidine (TMB) were obtained from Sigma (USA). Ovalbumin (OVA) and bovine serum albumin (BSA) were purchased from Beijing Dingguo Biotechnology Development Center (China). Nitrocellulose (NC) membranes were purchased from Millipore (USA). Other organic solvents were obtained from China National Medicines. An iMark Absorbance Microplate Reader (BIO-RAD, USA), BioDot ZX1000 dispensing platform with Frontline contact tips, and BioDot CM4000 guillotine cutter were used.
The Laboratory mice were purchased from Slac Laboratory Animal Company with Permit No SCXK 2008-0016. The Slac Laboratory Animal Company was initiated and established by the Chinese Academy of Sciences and specially engaged in the research, development, and business of experimental animals.
Synthesis of OA-BSA and OA-OVA conjugates
OA was conjugated to BSA or OVA by N-hydroxysuccinimide ester method as an immunogen or a coating antigen. The protocol was similar to the published procedure (Llamas et al. 2007), modified by using BSA as the carrier protein instead of bovine thyroglobulin.
Two mL OA solution, 2 mL BSA solution, and 2 mL OA-BSA solution, with PBS as a blank control, were scanned using a UV spectrophotometer in the 200–500-nm range to determine the maximum absorption peak. The coupling ratio of the OA and BSA can be estimated according to Yang et al. (1998), from that we can obtain the coupling result from the coupling ratio.
Hybridoma cell preparation for producing monoclonal antibody
Four BALB/c female mice at 8–10 weeks of age, with a body weight of approximately 18 g, were used. The initial immunization consisted of 150 μg OA-BSA conjugate and Freund’s complete adjuvant at the site of the foot pad. After 3 weeks, the secondary booster immunization consisting of 200 μg conjugate was administered with Freund’s incomplete adjuvant by subcutaneous injection. Subsequent booster injections were performed by intramuscular injection with 200 μg of conjugate and Freund’s incomplete adjuvant every 2 weeks. The mice were monitored by ELISA for specific antibody titer using tail bleeds taken 10 days after each booster. The most responsive mouse was selected after immunizations, and it received a final booster of 200 μg conjugate intraperitoneally only at 3 days prior to the fusion.
Splenocytes from the immunized mouse were mixed with myeloma cells (SP2/0) at a ratio of 5:1 and fused with 50 % PEG6000 as fusogenic reagent. The fused cells were cultured in DMEM containing 20 % HAT in the incubator at 37 °C. After 5 days, the hybridoma cell populations were replanted in DMEM containing HAT, and after 5–10 days, the hybridoma cell populations were replanted in MEM containing HT. The subsequent culture is in the DMEM.
The hybridoma cell population was then cloned three times by limiting dilution with the indirect competitive EIA. The cloned cells line was amplified in ascites, and the best cloned cell stored in liquid nitrogen. Monoclonal antibody was produced by the mouse ascitic method and purified by ammonium sulfate precipitation and affinity chromatography.
Establishment of idc-ELISA for OA detection
Effect of methanol concentration in OA solution on idc-ELISA
For the negative-control experiment (without OA), indirect ELISA was performed by mixing methanol at different final concentrations (range 0 to 50 %) with an equal volume of monoclonal antibody. The effect of methanol concentration on this immunoassay was assessed by the absorption values (OD450).
For the positive-control experiment, indirect competitive ELISA was performed by mixing methanol at different final concentrations (20, 30, and 40 %) and PBST with an equal volume of monoclonal antibody. The inhibition rate was calculated in order to study the effect of methanol concentration on idc-ELISA and to choose the best diluter. All the assays were performed in triplicate.
Generation of calibration curve for OA
A calibration curve for idc-ELISA was generated as follows: A total of 100 μL coating solution with OA-OVA at a concentration of 1.25 μg L−1 (diluted with 0.05 mol L−1 carbonate-buffered saline, pH 9.6) was added to the wells and incubated at 4 °C overnight. After incubation, drying, and washing each well for 5 min with PBST three times, buffers in the wells were removed. The blocking step was performed at 37 °C for 2 h with 200 μL 1 % gelatin. Subsequently, 50 μL McAb solution (1:10,000) and 50 μL of OA standard solution (OA concentrations: 200, 100, 50, 25, 12.5, 6.25, 5, 2.5, 1.25, 0.625, 0.3125, 0.156, and 0.078 ng mL−1 diluted with 0.05 mol L−1 PBS containing 0.05 % Tween-20, pH 7.4) were added together into the coated wells. After incubation at 37 °C for 1 h, the wells were washed with PBST and100 μL of diluted goat anti-mouse IgG-HRP solution (1:5,000) was added, and the mixture was incubated again at 37 °C for 1 h. After again washing the wells with PBST, 100 μL of TMB substrate solution was added into the wells, and the enzyme reaction was terminated after 10–15 min by adding 50 μL of 2 mol L−1 H2SO4 to each well. Measurement was by absorbance at 450 nm. The calibration curve was generated with log10 OA concentrations plotted along the x-axis and the inhibition rate plotted along the y-axis.
Cross-reactivity with other toxins (DTX-1, STX, MC-LR)
To determine the specificity, the cross-reactivity of the anti-OA monoclonal antibody with DTX-1, GYM, PTX2, SPX1, YTX, STX, and MC-LR was determined by indirect competitive ELISA. Toxin dilutions were made to provide a concentration range from 0 to 50 ng mL−1. Following the generation of a calibration curve for idc-ELISA, the cross-reactivity rate (CR) of the toxin (X) with OA based on 50 % inhibitory concentration (IC50) was calculated using the following equation:
Establishment of immunogold labeling assay method for OA detection
Preparation of colloidal gold suspension
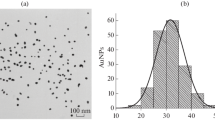
All glassware used in this preparation was thoroughly cleaned in aqua regia [HCl/HNO3 (3 v/v)], rinsed in Milli-Q purified water, and oven-dried prior to use. Colloidal gold particles (mean diameter 30 nm) were prepared by the sodium citrate method described by Frens (1973). Briefly, 100 mL of 0.01 % HAuCl4 solution (in Milli-Q purified water) was thoroughly boiled in a 250-mL conical beaker, and then 1.5 mL of 1 % trisodium citrate solution was added with constant stirring. After the color of solution changed to wine-red for about 45 s, it was boiled for another 5 min. After the heating source was removed, the colloidal gold solution was stirred for another 10 min. The solution was stored in a dark bottle at 4 °C and used to prepare colloidal gold-McAb conjugate as soon as possible. The dispersion and diameters of the colloidal gold particles were examined by a transmission electron microscope.
Preparation of colloidal McAb-gold conjugate
The pH of the colloidal gold solution for anti-OA Mcab conjugation was adjusted to pH 8.5 with 0.1 M K2CO3. With gently stirring, 0.2 mL of purified anti-toxin McAb (0.2 mg mL−1) was added drop by drop to 2 mL of the colloidal gold solution. The mixture was gently mixed for 1 h, blocked by 20 mg BSA for 30 min, and centrifuged at 7,825×g for 30 min. After centrifugation, the gold pellets were suspended in 100 μL dilution buffer [20 mM Tris/HCl buffer (pH 8.2) containing 1 % (w/v) BSA] and then stored at 4 °C before the next step.
Preparation of immunochromatographic test strips and assay of OA
Control and test lines were sprayed onto a NC membrane (25 × 300 mm). For the test line, 0.75 mg mL−1 of OA-OVA conjugate and for the control line 1.0 mg mL−1 of goat anti-mouse IgG were applied at 0.5 μL cm−1. Membranes were dried at 25 °C overnight, blocked with 1 % (w/v) BSA in 0.05 M PBS for 30 min, and washed three times with 0.05 M PBS for 5 min and again dried overnight. Nitrocellulose membrane and wick pad were assembled on an adhesive backing card and strips of 5 mm width were cut and then stored at 4 °C before use.
The assay was carried out by applying 70 μL of 25, 12.5, and 6.25 ng mL−1 OA standard solution to the bottom of the device. After 3 min, the results were visually evaluated.
Preparation of sample and toxin extraction
Samples were prepared according to Llamas et al. (2007). Whole mussel tissues were removed from the shell, drained in a sieve (5 min), and homogenized (1 min) in a blender. The homogenates, when not used immediately, were stored at −20 °C.
The homogenized mussel sample (5 g) was put into a centrifuge tube and spiked with 1 mL OA standard solution. Methanol (20 mL) was added to the sample and mixed (30 min) and centrifuged (2,300×g, 10 min). The supernatant was placed in a dry glass bottle and evaporated to dryness at 45 °C in a water bath. The final residue was dissolved in methanol (1 mL) and washed with hexane (1 mL), and the methanol layer was then dried as previously described. The second resulting residue redissolved in methanol (100 μL) was subsequently 40 times diluted with PBS buffer (containing 0.05 % Tween-20), mixed (10 s) and filtered through a 0.45-μm filter membrane. The extraction was tested by idc-ELISA and immunochromatographic strips. For immunochromatographic strip test, the extraction solution must be diluted 40 times with PBS buffer.
Results
Conjugation between OA and BSA
Figure 1 shows the result of the conjugations between OA and BSA. The absorption curve and peak for OA-BSA conjugating products were different from those of BSA and OA alone. The absorption peaks of OA and BSA were at 260 nm and 280 nm, respectively, while OA-BSA had an absorption peak at 262 nm similar to OA. There were two peaks for BSA, and there were two peaks in the absorption spectrum of OA-BSA conjugating products, very similar to BSA. The second peak of the conjugate was shifted toward the OA peak at 262 nm, twice the amplitude of the BSA peak at 280 nm. Thus, the immunogen was synthesized successfully. An estimated molar coupling ratio was 18:1.

A The UV spectrum of OA; B the UV spectrum of OA-BSA; C the UV spectrum of BSA
Cell hybridization and screening of cell line for high producing McAbs
The mouse with highest titers was selected to hybridize. Fusion cells were inoculated in nine enzyme-labeled plates. After 6 days, 850 wells had cell growth with the fusion rate at 98.3 %. By the indirect ELISA method selection, there were 17 positive wells, four of them strong. The positive rate was 3.65 %. After three times cloning and identification, the clone cell 3H4 was selected which can also secrete the antibody in a stable manner after subculture, frozen storage, and recovery. The result with indirect ELISA showed that the titers of culture supernatants and ascites were 1:640 and 1:1.28 × 106, respectively.
Effect of methanol concentration on ELISA detection
Figures 2 and 3 show the effect of methanol concentration on ELISA, and the maximum absorbance (without toxin present) value for ELISA results varied at different methanol concentrations. Absorbance values steadily increased with increasing methanol concentrations above 10 % (Fig. 2). By using indirect competitive ELISA, the inhibition rate reduced with increased methanol concentration (Fig. 3). Consequently, during the step of “Sample preparation and toxin extraction,” the second resulting residue redissolved in methanol (100 μL) was subsequently diluted in 900 μL of PBS buffer to reduce the methanol concentration below 10 %.

Effect of methanol concentration on ELISA absorbance

Effect of OA diluter (methanol) concentration on idc-ELISA
Determination of ELISA sensitivity for the limit of detection
Figure 4 shows the OA calibration curve of idc-ELISA. The calibration curve for OA had a good linearity over the concentration range of 0.3125–50 ng mL−1, with a limit of detection (LOD) of 0.45 ng mL−1 (thrice the standard deviation at zero dose) and the IC50 against OA was 2.59 ng mL−1.

OA calibration curve of idc-ELISA inhibition rate = (A 0 − A)/A 0 × 100 % (A is the absorbance of the well containing OA; A 0 is the absorbance of the well without OA)
Examination of cross-reactivity for the McAb against OA
The cross-reactivity of the OA antibody with DTX-1, GYM, PTX2, SPX1, YTX, five analogs of OA in DSP, STX, as well as MC-LR was tested. The results are shown in Table 1, and the cross-reactivity rate of the antibody with DTX-1 was 53.1 %, while there was no cross-reactivity with GYM, PTX2, SPX1, YTX, STX, or MC-LR. Because DTX-2 and other DTXs cannot be obtained in China, we could not determine the cross-reactivity between the McAb against OA and DTXs. Related supplementary experiments will be done when these toxins can be obtained.
Determination of the detection limit for immunochromatographic test strips
Various concentrations of OA standard solution (0, 1.56, 3.125, 6.25, 12.5, 25, 50, 500, 1,000, and 5,000 ng mL−1) were assayed using the immunochromatographic test strip (Table 2; Fig 5). When OA concentration was from 0 to 3.125 ng mL−1, the test lines in the test strips all presented red color. At toxin concentrations ranging from 6.25 ng mL−1 to 5,000 ng mL−1, the test lines did not present red color. This indicates that the LOD for the test strip was 3.12 ng mL−1 and 6.25 ng mL−1.

Immunochromatographic detection of OA using test strip
Recovery of OA in mussel samples
The recoveries of OA from mussel samples added with known amount of OA (250, 500, 1,000, and 2,000 ng mL−1) were determined by idc-ELISA (Table 3). Over the mussel sample OA content range of 250 to 2,000 ng mL−1, the recoveries of OA ranged from 88.03 to 96.26 %, and average of recovery was 92.76 % with good reproducibility with a coefficient of variation (CV) of 2.41-6.76 %.
Detection of OA in mussel samples by test strip
According to the US Federal Drug Administration (FDA), the critical OA content in mussels is 20 μg per 100 g (= 1,000 ng per 5 g). When all of the OA was extracted from 5 g mussel with 1 mL PBS or methanol, the OA concentration was just 1,000 ng mL−1. This OA concentration was too high for testing by test strips because the LOD is 3.125–6.25 ng mL−1, and so the solution sample needs be diluted. As the average recovery of OA from mussel was about 93.94 %, the dilution multiple therefore is:
According to the sample processing method in the “Materials and methods,” 250, 500, 1,000, and 2,000 ng mL−1 solutions were extracted from mussels and diluted 150 times, and these samples were tested with the immunochromatographic test strips (Table 3). When the OA concentration in the solution samples ranged from 250 to 500 ng mL−1, the test line was red, indicating that the mussel tissue was safe and edible. When the OA concentration in the samples was higher than 1,000 ng mL−1 (above 6.00 ng mL−1, diluted 150 times), the test line showed no color, indicating that the mussel tissue was not safe.
Detection of OA in mussel samples fed with Prorocentrum lima
The OA contents in mussels fed with P. lima for 0–384 h were assayed by both ELISA and test strips (Table 4). With test strip detection, the test lines in test strips for mussels fed with P. lima for 0 and 96 h where the OA concentrations were lower than 6.25 ng mL−1, were red, which indicated that the mussels were safe, whereas the test lines for mussels fed with P lima from 96 to 384 h (OA concentration was higher than 6.25 ng mL−1) showed no red color, indicating that the mussels were not safe to eat.
Discussion
Currently, various techniques have been developed to detect toxins such as OA, and the most widely used method is the mouse bioassay (Yasumoto et al. 1978). This method had several limitations including non-specific toxin detection, poor quantification, risk of false positives caused by fatty acids, and low reproducibility (Dennison and Anderson 2007). A number of alternative methods have been proposed such as cytotoxicity assays (Amzil et al. 1992), protein phosphatase assays (Luu et al. 1993; Honkanen et al. 1996; Simon and Vemoux 1994; Minoru et al. 1995), LC coupled with fluorescence detection, and LC coupled with mass spectrometry (James et al. 2000; Mouratidou et al. 2006; Mendoza et al. 2008). However, these methods are time-consuming and require expensive instruments and trained personnel. Of these methods, immunoassay has become one of the most popular methods for detecting toxins in shellfish due to its high sensitivity, simplicity, and small sample volume required.
In this study, we first established an indirect competitive enzyme-linked immunosorbent assay for OA detection in 2009 successfully. We obtained an antibody against OA with highly sensitivity and specificity: the IC50 against OA was 2.59 ng mL−1, and the detection limit was 0.45 ng mL−1. These results suggest that this method is more sensitive than reported immunological methods (Shestowsky et al. 1992; Matsuura et al. 1994; Núnez and Scoging 1997; Laycock et al. 2006; Llamas et al. 2007; Liu et al. 2008; Dubois et al. 2010).
Secondly, we developed a competitive immunochromatographic assay based on the colloidal gold-labeled monoclonal antibody against OA. This method is easy and fast (3–5 min) with a test sensitivity of 6.25 ng mL−1 which is lower than the results of others (Stewart et al. 2009). It provides a preliminary semi-quantitative result on whether the toxin concentration in the sample is higher than the detection limit. Visual results of the test are in good agreement with ELISA results. Other approaches, such as for example cytotoxicity assay and LC-MS/MS methods, could provide unique insights into the presence of OA. Although the immunogold assay is not as accurate and its sensitivity is lower than these techniques, it is easy, rapid, and convenient to apply, requiring no animals and equipment, and can be finished within 3–5 min without complicated handling procedures. Thus, in terms of the overall speed and simplicity, this method is superior to other methods, including immunoassays. Our data suggest that this immunochromatographic strip may be useful as a rapid screening method in the field and for environmental monitoring with a period of no more than 40 min for extraction, dilution, and detection.
Although OA antibody preparation has been reported (Usagawa et al. 1989; Stewart et al. 2009), an OA ELISA detection method (Dubois et al. 2010) and a biosensor detection method (Stewart et al. 2009) have been established, and commercial ELISA test kits (DSP-check ELISA test kit from UBE Industries, Tokyo, Japan; Rougier Bio-Tech ELISA test kit) are available, there is no commercial immunochromatographic strip test currently available.
The FDA of the USA proposed a maximum limit for OA in shellfish meat of 0.2 mg kg−1 (Burgess and Show 2001). In European Union countries, an ideal assay should show a positive result at or above a regulatory limit of around 0.2–0.6 mg OA per kg of tissue but a clear negative below this concentration (Burgess and Show 2001). In China, the maximum limit for OA in shellfish meat is 0.8 mg kg−1 (Zhang and Zhang 2011). Therefore, the rapid strip had a great application potential for OA detection, especially for the purpose of food safety.
References
Amzil Z, Pouchus YF, Boterff JL, Roussakis C, Verbist JF, Marcaillou-Lebaut C, Masselin P (1992) Short-time cytotoxicity of mussel extracts: a new bioassay for okadaic acid detection. Toxicon 30:1419–1425
Burgess V, Show G (2001) Pectenotoxins—an issue for public health: a review of their comparative toxicology and metabolism. Environ Int 27:275–283
Campàs M, Iglesia PDL, Berre ML, Kane M, Diogène J, Marty JL (2008) Enzymatic recycling-based amperometric immunosensor for the ultrasensitive detection of okadaic acid in shellfish. Biosens Bioelectron 24:716–722
Carmody EP, James KJ, Kelly SS (1996) Dinophysistoxin-2: the predominant diarrhoetic shellfish toxin in Ireland. Toxicon 34:351–359
Dennison N, Anderson DB (2007) The 3 “R”s approach to marine biotoxin testing in the UK. In: Proc. 6th World Congress on Alternatives & Animal Use in the Life Sciences, AATEX 14, Special Issue, Tokyo, Japan, pp 757–761
Dubois M, Demoulin L, Charlier C, Singh G, Godefroy SB, Campbell K, Elliott CT, Delahaut P (2010) Development of ELISAs for detecting domoic acid, okadaic acid, and saxitoxin and their applicability for the detection of marine toxins in samples collected in Belgium. Food Addit Contam 27:859–868
Frens G (1973) Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature Phys Sci 24:120–122
Garthwaite I (2000) Keeping shellfish safe to eat: a brief review of shellfish toxins, and methods for their detection. Trends Food Sci Tech 11:235–244
Honkanen RE, Mowdy DE, Dickey RW (1996) Detection of DSP toxins, okadaic acid and dinophysistoxin-1 in shellfish by serine/threonine protein phosphatase assay. J AOAC Int 79:1336–1343
James KJ, Bishop AG, Carmody EP, Kelly SS (2000) Detection methods for okadaic acid and analogues. In: Botana LM (ed) Seafood and freshwater toxins: pharmacology physiology and detection. CRC, New York, pp 217–238
Laycock MV, Jellett JF, Easy DJ, Donovan MA (2006) First report of a new rapid assay for diarrhetic shellfish poisoning toxins. Harmful Algae 5:74–78
Liu RY, Chen BJ, Liang YB, Xu DY, Liang B (2008) Preparation and application of the monoclonal antibody against okadaic acid. J Hyg Res 37:443–445 (in Chinese)
Llamas NM, Stewart L, Fodey T, Higgins CW, Velasco MLR, Botana LM, Elliott CT (2007) Development of a novel immunobiosensor method for the rapid detection of okadaic acid contamination in shellfish extracts. Anal Bioanal Chem 389:581–587
Luu HA, Chen DZX, Magoon J, Worms J, Smith J, Holmes CFB (1993) Quantification of diarrhetic shellfish toxins and identification of novel protein phosphatase inhibitors in marine phytoplankton and mussels. Toxicon 31:75–83
Matsuura S, Kita H, Takagaki Y (1994) Specificity of mouse monoclonal anti-okadaic acid and antibodies to okadaic acid and its analogs among diarrhetic shellfish toxins. Biosci Biotech Biochem 58:1471–1475
Mendoza WG, Mead RN, Brand LE, Shea D (2008) Determination of brevetoxin in recent marine sediments. Chemosphere 73:1373–1377
Minoru I, Yukiteru S, Takeharu I, Ikuko IO, Yoshiaki T, Yoshiko N, Akira T (1995) New analysis method for protein phosphatase type 2a inhibitors using the firefly bioluminescence system. Biosci Biotech Biochem 59:2235–2238
Morton SL, Tindall DR (1996) Determination of okadaic acid content of dinoflagellate cells: a comparison of the HPLC-fluorescent method and two monoclonal antibody ELISA test kits. Toxicon 34:947–954
Mouratidou T, Kaniou-Grigoriadou I, Samara C, Kouimtzis T (2006) Detection of the marine toxin okadaic acid in mussels during a diarrhetic shellfish poisoning (DSP) episode in Thermaikos Gulf, Greece, using biological, chemical and immunological methods. Sci Total Environ 366:894–904
Murakami Y, Oshima Y, Yasumoto T (1982) Identification of okadaic acid as a toxic component of a marine dinoflagellate Prorocentrum lima. Bull Jpn Soc Sci Fish 48:69–72
Murata M, Shimatani M, Sugitani H, Oshima Y, Yasumoto T (1982) Isolation and structural elucidation of the causative toxin of diarrhetic shellfish poisoning. Bull Jpn Soc Sci Fish 48:549–552
Núnez PE, Scoging AC (1997) Comparison of a protein phosphatase inhibition assay, HPLC assay and enzyme-linked immunosorbent assay with the mouse bioassay for the detection of diarrhetic shellfish poisoning toxins in European shellfish. Int J Food Microbiol 36:39–48
Quilliam MA (2003) The role of chromatography in the hunt for red tide toxins. J Chromatogr A 1000:527–548
Shestowsky WS, Quilliam MA, Sikorska HM (1992) An idiotypic–anti-idiotypic competitive immunoassay for quantitation of okadaic acid. Toxicon 30:1441–1448
Simon JF, Vemoux JP (1994) Highly sensitive assay of okadaic acid using protein phosphatase and paranitrophenyl phosphate. Nat Toxins 2:293–301
Stewart LD, Elliott CT, Walker AD, Curran RM, Connolly L (2009) Development of a monoclonal antibody binding okadaic acid and dinophysistoxins-1,-2 in proportion to their toxicity equivalence factors. Toxicon 54:491–498
Souto ML, Fernández JJ, Norte M, Fernández ML, Martinez A (2001) Influence of amino acids on okadaic acid production. Toxicon 39:659–664
Tachibana K, Scheuer PJ, Tsukitani Y, Kikuchi H, Engen DV, Clardy J, Gopichand Y, Schmitz FJ (1981) Okadaic acid, a cytotoxic polyether from two marine sponges of genus Halichondira. J Am Chem Soc 103:2469–2471
Usagawa T, Nishimura M, Itoh Y, Uda T, Yasumoto T (1989) Preparation of monoclonal antibodies against okadaic acid prepared from the sponge Halichondria okadai. Toxicon 27:1323–1330
Yang LG, Wei PH, Guo AZ (1998) The enzyme immunoassay. Nanjing University Press, Nanjing, pp 279–281 (in Chinese)
Yasumoto T, Oshima Y, Yamaguchi M (1978) Occurrence of a new type of shellfish poisoning in the Tohoku District. Bull Jpn Soc Sci Fish 44:1249–1255
Zhang WB, Zhang ZJ (2011) The establishment of national food safety standards in the aquatic food shellfish toxins indicators. China Health Stand Manage 2(6):37–41 (in Chinese)
Acknowledgments
This study was supported by the China National Marine 863 Program (2007AA09Z406), the Project for Excellent Academic Leaders (08SD14037), the International Cooperation Project (08540702600), the Biomedical Sciences Project (10391901900), and the Mountaineering Project (06dz12015) from Shanghai Committee for Science and Technology, the Hydrobiological Project of Shanghai Leading Academic Discipline (III) (S30701), China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, L., Liu, J., Wang, Q. et al. Development of an immunochromatographic strip test for the rapid detection of okadaic acid in shellfish sample. J Appl Phycol 25, 1091–1099 (2013). https://doi.org/10.1007/s10811-012-9949-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-012-9949-3