
Abstract
Polysaccharides, galactans, obtained from edible red seaweed Hypnea musciformis were characterized by molecular weight and infrared spectroscopy analysis and were evaluated for antioxidant activity in vitro and for their effects on cell viability. The main components were galactose and sulfate presenting low protein contamination. These sulfated galactans (F1.0) showed a polydisperse profile, and signs in infrared analysis were attributed to a sulfate ester S = O bond, the presence of a 3,6-anhydrogalactose C–O bond, nonsulfated β-d-galactose, and a C–O–SO4 bond in galactose C4. The NMR analysis showed signals at about 95 and 92 attributed to anomeric carbon of 4-linked 3,6-anhydro-α-d-galactopyranose residue of κ-carrageenans and 4-linked 3,6-anhydro-α-d-galactopyranose2-sulfate of ι-carrageenans. Sulfated galactan F1.0 showed strong antioxidant activity under lipid peroxidation assay where F1.0 at 8 mg mL−1 promoted 57.92% peroxidation inhibition and displayed the scavenging activity on hydroxyl radicals in a dose-dependent manner leading to 32.5% scavenging of these radicals when 5 mg mL−1 of sulfated galactan F1.0 was used. The sulfated galactan fraction also exhibited strong inhibition on the H2O2-induced hemolysis model. Sulfated galactan F1.0 displayed low cytotoxic action in 3 T3 cells and moderate antitumoral action in HeLa cells. These results suggest that sulfated galactan F1.0 from H. musciformis has antioxidant potential, which is a great effect for a compound used as food and in the food industry.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Marine seaweeds are one of the major sources of biologic active compounds in nature (O´Sullivan et al. 2010). Among these compounds are the sulfated polysaccharides, which are polymers that present several effects on the biological process such as anticoagulant (Medeiros et al. 2008; Mao et al. 2009), antithrombotic (Fonseca et al. 2009), antiproliferative (Costa et al. 2010), antiviral (Ohta et al. 2009), antitumor (Lins et al. 2009), immunostimulating (Lee et al. 2010), anti-inflammatory (Zubia et al. 2007; Medeiros et al. 2008), and antioxidant action (Matsuhiro et al. 2005; Wang et al. 2008; Hu et al. 2010; Barahona et al. 2011).
Sulfated polysaccharides have a complex and heterogenous structure, and they function as structural components of the extracellular matrix of these organisms. Since 1967, Percival and McDowell (1967) have also claimed that polysaccharides from red seaweed could be sulfated galactans. These are sulfated polysaccharides, which generally show high molecular weight (≥100 kDa) and high electronegativity due to sulfated esters in the structure that make them fairly anionic, enabling electrostatic interaction with specific proteins leading to biologic actions (Usov et al. 1980; Pomin 2009; Silva et al. 2010). Sulfated galactans show residues with a linear structure of alternating 3-linked β-d-galactopyranose (D configuration) and 4-linked α-galactopyranose, which are in a D configuration in carrageenans and L configuration in agar. Some of the residues are in anhydrogalactose (Knutsen et al. 1994).
These polymers have a wide application in the food industry as sulfated galactans are hydrocolloids capable of forming aqueous gels (Lahaye 2001; Tuvikene et al. 2008), and because of this, they are used as a texturing agent with thickening and gelling properties for foods (McHugh 2003) and as stabilizers. In processed meats, such as, ham and turkey breast, carrageenans function as a water ligand agent preventing wet loss during cooking and therefore avoiding the undesirable dry texture of meats (Bixler and Porse 2011).
The red seaweed, Hypnea musciformis, is a natural source of kappa carrageenan, a commercially important galactan and therefore has been fairly exploited on the northeastern coast of Brazil (Oliveira 1998).
The normal metabolic process as well as the exogenous factors promotes oxygen reactive species generation which can induce pathologic effects such as carcinogenesis, atherosclerosis, DNA damage, and the degenerative process related to aging (Liu et al. 1997; Mau et al. 2002). An antioxidant agent slows or prevents oxidizable cellular substrate oxidation, leading to oxygen reactive species generation inhibition or scavenging. Some synthetic antioxidant agents such as BHA, BHT, and TBHQ are used mainly in processed meat; however, due to safety concerns about using these agents, there is an increasing interest in the search for natural antioxidants (Formanek et al. 2001).
In their natural habitat, marine seaweeds are exposed to high oxygen concentrations and light in a combined manner, promoting free radical generation just as other oxidizing agents do. Therefore, seaweeds do not suffer any photodynamic damage, which suggests the presence of antioxidant components and mechanisms in these organisms (Matsukawa et al. 1997). The aim of this study was to evaluate the in vitro antioxidant activity of sulfated galactans from H. musciformis.
Materials and methods
Polysaccharide fraction 1.0 (F1.0) extraction
The red seaweed was collected on the southern coast of Rio Grande do Norte State, Búzios Beach, Brazil. It was washed in flowing water, dried, crushed, and weighed in our laboratory. To the dried seaweed were added two volumes of acetone to remove the lipids. Then the acetone was removed, and the seaweed was dried one more time at room temperature obtaining cetonic powder. To this powder were added NaCl 0.15 M (two volumes) and the proteolytic enzyme maxatase (pH 8.0). The suspension was filtered and a supernatant named polysaccharides crude was acquired, which was then centrifuged at 8.000 × g for 15 min. After that, the precipitate was discarded and the supernatant was fractionated with increasing volumes of acetone and centrifuged (Dietrich et al. 1995). Sulfated galactan fraction 1.0 (F1.0) was precipitated with one volume of acetone.
Chemical composition
Total sugars were quantified by phenol-H2SO4 reaction according to Dubois et al. (1956) using d-galactose as standard. Sulfate content was measured after acid hydrolysis (HCl 6 N, 6 h, 100°C) by the turbidimetric method (Dodgson and Price 1962). Protein content was determined by the Bradford (1976) method using Coomassie Brilliant Blue reagent and bovine serum albumin as standard.
Phenolic compounds were measured by the Folin Ciocalteau method (Swain and Hills 1959) with few modifications using 0.5 mL ethanol, 2.5 mL distilled water, 0.25 mL Folin Ciocalteau reagent, 0.5 mL sodium carbonate, and 0.5 mL sulfated polysaccharides from H. musciformis (1 mg mL−1). A reagent blank was prepared using distilled water. Absorbance was determined at 750 nm against the blank, and a gallic acid calibration curve (0–500 mg L−1) was constructed and was used to determine the total phenolic content of the samples, expressed as gallic acid equivalents.
Molecular weight determination
Molecular weight was determined by gel filtration chromatography Sephadex G-100 gel column (1.2 × 100 cm). The column was eluted with 0.1 M NaCl at 1.2 mL/ 10 min/tube. The sulfated galactan fraction 1.0 was assayed for sugar content using the phenol–sulfuric acid method with d-galactose as standard (Dubois et al. 1956) and protein content (Bradford 1976). The column was calibrated with standard Dextran from Pharmacia (molecular weight, 10,000; 40,000; 70,000; 133,000; 482,000; and 2,000,000 Da). A standard curve was established before sample analysis.
Infrared and NMR spectroscopy
The IR spectrum of the sulfated galactan fraction 1.0 from H. musciformis was carried out using a Fourier transform infrared spectrophotometer (FTIR, Bruker, Germany) equipped with OPUS 3.1 software. The fraction was ground with KBr powder and then pressed into pellets for FTIR measurement in the frequency range of 4,000–500 cm−1.
13C determinations were carried out using a 200-MHz Bruker model DRX Avance spectrometer incorporating Fourier transform. Samples were dissolved in H2O-d and examined at 70 °C.
Antioxidant activity assays
Hydroxyl radicals scavenging was determined by a modified Smirnoff and Cumbes (1989) method. Phenanthroline, 0.5 mL (5 mM L−1), 0.75 mL phosphate buffer (20 mM; pH 7.4), and 0.5 mL Fe2SO4 (7.5 mM L−1) were added to 0.5 mL of different concentrations of sulfated galactan F1.0 (0.08–5 mg mL−1). Then 0.5 mL 15% H2O2 was added and the mixture was incubated at 37 °C for 90 min. After incubation, the mixture was centrifuged at 1,000 × g for 5 min and absorbance was measured at 536 nm. The scavenging activity on hydroxyl radicals was calculated using the following equation: scavenging activity (%) = (1 − A sample/A control) × 100, where A control is absorbance of the mixture without samples and A sample is absorbance of the mixture with samples.
Ferrous ion chelating activity was determined using different concentrations of sulfated galactan F1.0 (0.08–5 mg mL−1) mixed with 0.025 mL FeCl2 2 mM and incubated for 1 min at room temperature. Ferrozine, 0.1 mL, 5 mM was added and the mixture was incubated for 20 more minutes at the same temperature. Next, distilled water was added to reach 2 mL, and absorbance was measured at 562 nm. The chelating activity percentage was given by the following formula: activity rate (%) = [(A positive control − A sample)/A positive control] × 100, where A positive control is positive control absorbance (sample is replaced by distilled water) and A sample refers to sample absorbance (Saltarelli et al. 2009).
The scavenging activity on 1,1-diphenyl-2-picrylhydrazyl (DPPH) radicals was determined according to the Ye et al. (2008) method with slight modifications. Different concentrations of 0.1 mL of sulfated galactan F1.0 (0.08–5 mg mL−1) was added to the 1.5 mL ethanol solution 0.1 mM of DPPH. After 30 min at room temperaure, the absorbance was measured at 517 nm. The scavenging activity on DPPH radicals was calculated using the following equation: scavenging activity (%) = (1 − A sample/A control) × 100, where A control is absorbance of the ethanol solution of DPPH without sample (sample was replaced by ethanol) and A sample is absorbance of the ethanol solution of DPPH with tested samples.
Superoxide radicals were generated in the phenazin methosulphate–NADH system with 3 mL of Tris–HCl buffer (16 mM; pH 8) containing 50 μM nitroblue tetrazolium, 10 μM phenazin methosulphate, 78 μM NADH (reduced form), and different concentrations of sulfated galactan F1.0 (0.08–5 mg mL−1). Colorimetric reaction was detected at 560 nm (Zhang et al. 2003). Percentage of scavenging activity on superoxide radicals was determined by the formula: (%) = [(A positive control − A sample)/A positive control − A negative control)] × 100, where A positive control is positive control absorbance (all reagents without sample), A sample is sample absorbance, and A negative control is negative control absorbance (NADH absence).
Reducing power was evaluated based on Yuan et al. (2005) with slight modications. Sulfated galactan, 0.2 mL, F1.0 in different concentrations (0.08–5 mg mL−1) was incubated with 0.5 mL phosphate buffer (0.2 M; pH 6.6) and 0.5 mL K3Fe(CN)6 (1.0% w/v) in a water bath for 20 min at 50°C. Samples were cooled and mixed with 0.5 mL TCA (10% w/v) and were then centrifuged for 10 min at 3,000 × g. The supernatant (0.5 mL) was mixed with 0.1 mL FeCl3 solution (1.0%, p/v) and 0.5 mL distilled water. Absorbance was measured at 700 nm, and the results were expressed in absorbance. The greater the absorbance, the greater the reducing power. As a negative control, a buffer was used instead of the sample and as a reaction blank all reagents were used with the exception of ferric chloride, which was replaced by water. BHT was used as standard antioxidant.
Lipid peroxidation inhibition was determined by acid thiobarbituric reaction using egg yolk as an oxidable substrate. The system was generated with 0.25 mL homogenized egg yolk in 10% PBS (0.2 M; pH 7.4), 0.025 mL FeSO4 0.07 M (to start lipid peroxidation), and 0.25 mL of sulfated galactan F1.0 in different concentrations (0.08–5 mg mL−1). The mixture was incubated at 37°C for 30 min. After incubation, 0.75 mL 20% (v/v) trichloroacetic acid and 0.75 mL 0.8% (w/v) thiobarbituric acid were added. Then the mixture was shaken and heated at 100°C for 15 min and centrifuged at 2,000 × g for 10 min and measured at 532 nm (Zhang and Yu 1997). Percentage of inhibition of lipid peroxidation was expressed in inhibition rate (%) = [1 − (A sample/A positive control)] × 100, where A sample and A positive control refers to sample and positive control absorbance (sample absence), respectively.
H2O2-induced hemolysis evaluation was by the Ugartondo et al. (2009) method with slight modifications. A 10% red blood cell suspension was added to increasing concentrations of sulfated galactan F1.0 (diluted in saline solution). Then 400 mM L−1 H2O2 was added, and the mixture incubated at 37°C for 90 min on a shaker. Erythrocytes were incubated with only saline as the negative control. In the positive control, hemolysis was induced by H2O2 but no treatment with sulfated galactan fraction 1.0 was done. After incubation, the erythrocytes were centrifuged at 1,000 × g for 5 min, and the supernatant absorbance measured at 540 nm. Results are expressed in hemolysis induced by H2O2 inhibition percentage (%) = (1 − (A sample − A negative control)/(A positive control − A negative control) × 100, where A sample, A negative control e A positive control refer to tested samples, negative control, and positive control absorbance, respectively.
Cytotoxic action of F1.0 on different ABO and Rh groups of red blood cells
Direct hemolytic assay was as described by Belokoneva et al. (2003) with slight modifications. Blood samples were collected in EDTA tubes. Red blood cells were rinsed with PBS buffer (0.15 M; pH 7.4) until the supernatant reached a transparent state. A 10% red blood cell suspension in PBS buffer was incubated with different amounts of sulfated galactan F1.0 (50, 100, 150 μg) at room temperature for 1 and 6 h. After incubation, red blood cell suspension was centrifuged at 1,000 × g for 5 min and supernatants were read at 540 nm. As positive and negative control, the red blood cell suspension was incubated under the same conditions with 1% Triton X-100 and PBS buffer, respectively. Results of direct hemolytic assay were expressed as hemolysis percentage (%) = (A sample − A PBS)/(A 1% Triton X-100 − A PBS) × 100, where A sample, A PBS, and A 1% Triton X-100 refers to tested samples, negative control (PBS), and positive control (1% Triton X-100), respectively. Incubation was done in triplicate using different groups of red blood cells (A, B, AB, and O groups) as well as of different Rh groups (A positive and A negative red blood cells).
Effect of F1.0 on cell viability (MTT assay)
The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) method is a cellular viability assay used to evaluate the cytotoxicity of compounds. The cytotoxicity of sulfated galactan F1.0 was measured as previously described by Mosmann (1983) using HeLa and 3 T3 cell lineage. Cultures were exposed to 25, 50, and 100 μg of sulfated galactan F1.0 (triplicate) and incubated at 37°C for 24, 48, and 72 h. After incubation, 100 μL of medium containing MTT (final concentration, 5 mg mL−1) was added in each well, and plates were incubated at 37°C for 4 h. The supernatant was removed, and ethanol P.A. was added to each well. The plate was mixed thoroughly to dissolve the dark blue crystals and was then read at 570 nm.
Statistical analysis
Values are presented as mean ± SEM. Analysis of variance (ANOVA) and the Turkey–Kramer test were used for biological activity data assessment, with p < 0.05 accepted as statistically significant. EC50 and IC50 values were calculated using GraphPad Prism 5.
Results
Crude polysaccharides were extracted from H. musciformis and then fractionated with increasing concentrations of acetone. From this fractionating the sulfated galactan fraction 1.0 (F1.0) was obtained, which was precipitated with one volume of acetone.
The chemical composition of sulfated galactan F1.0 is presented in Table 1. The results show that sulfated galactan F1.0 is mainly composed of carbohydrates (49.70 ± 0.10%) and high sulfate content (44.59 ± 0.015%). However, it possesses a low protein contamination (0.92 ± 0.001%) and low phenolic compounds content (4.79 ± 0.016%). These results indicate that the total phenolics and protein contents were removed in the extraction process.
Gel filtration chromatography of sulfated galactan F1.0 monitored by total sugar content (Dubois et al. 1956) and protein content (UV light/280 nm) showed a polydisperse profile. Sulfated galactan F1.0 is composed of two molecular weight populations of polysaccharides of MW 147–155 kDa. This sulfated galactan (F1.0) showed a low contamination by proteins.
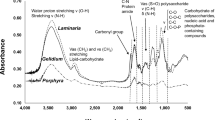
Infrared analysis of sulfated galactan F1.0 extracted from H. musciformis showed bands in 3,423; 2,930; 1,653; 1,416; 1,262; 1,156; 1,074; 930; 900; and 850 cm−1 (Fig. 1). The band in 1,262 cm−1 was from sulfate esters S = O bond, which refers to the presence of sulfation in the sample (Pereira et al. 2009). This signals intensity was positively related to the sulfate content of the tested compound (Hu et al. 2010). Signals in 1,074 and 930 cm−1 revealed the presence of a 3,6-anhydrogalactose C–O bond. Signs in 900 and 850 cm−1 were attributed to non-sulfated β-d-galactose and a C–O–SO4 bond in galactose C4, respectively (Pereira et al. 2009). Bands in 3,423 and 2,930 cm−1 were from stretch vibration of H–O existing in the hydrogen bond of molecules and stretch vibration of –CH, respectively (Sun et al. 2009).

Infrared spectroscopy analysis of F1.0 polysaccharides from H. musciformis
F1.0 NMR analysis (Fig. 2) showed signals at about 95 and 92 ppm attributed to anomeric carbon of 4-linked 3,6-anhydro-α-d-galactopyranose residue of κ-carrageenans and 4-linked 3,6-anhydro-α-d-galactopyranose 2-sulfate of ι-carrageenans (Usov and Shashkov 1985). The signal at 97.0 ppm may probably be attributed to C-1 of 3,6-anhydrogalactose residues in oversulfated carrabiose units having sulfate at C-2 or C-6 of 3-linked β-d-galactose residues (Van de Velde et al. 2002). Signal at 102.9 ppm belongs to the anomeric carbon of 3-linked β-d-galactose residues and is common to all the 3,6-anhydrogalactose-containing carrageenans.

13C-NMR analysis of F1.0 obtained from Hypnea musciformis
Antioxidant activity assays
Hydroxyl radicals, subsequently, radicals are the most prejudicial reactive species and the ones that cause more oxidative damage in biomolecules (Ke et al. 2009). Removal of these radicals is very important for antioxidant defense in cells and in the food system (Aruoma 1998). Sulfated galactan F1.0 displays the scavenging activity on hydroxyl radicals in a dose-dependent manner leading to 32.5% scavenging of these radicals when 5 mg mL−1 of F1.0 was used, as shown in Table 2. The EC50 was 13.68 mg mL−1.
Iron has high reactivity and is known as the most important prooxidant to lipid peroxidation. The ferrozine forms a complex with ferrous ion (Fe+2); however, in the presence of iron chelating agents, there is an interruption of this complex formation which can be evaluated by the decreasing of red color generated by the complex. Therefore, color reduction refers to the chelating activity of the tested compound (Qi et al. 2006). In tested concentrations, sulfated galactan F1.0 displays low iron chelating activity with 8% of chelating activity at 5 mg mL−1 and with EC50 of 56.19 mg mL−1 (Table 2).
DPPH is a stable free radical that has been widely applied in antioxidant test models to evaluate the ability of natural compounds to scavenge DPPH radicals (Chen et al. 2008). In the presence of a hydrogen donator, the DPPH radical is reduced to diphenyl-pricryl-hydrazine, which has a yellow color leading to decreasing absorbance at 517 nm. Thus, the antioxidant activity of a given compound is expressed as the scavenge ability on DPPH free radicals (Wang et al. 2010). As shown in Table 2, sulfated galactan F1.0 displays the scavenging activity on DPPH of 9.88% when used at 5 mg mL−1 with an EC50 of 37.46 mg mL−1.
Superoxide radicals are usually the first radicals to be generated in cellular oxidation reactions (Liu and Ng 2000) and function as the precursor of singlet oxygen and hydroxyl radicals; thus, they are responsible for generating other oxidant agents (Athukorala et al. 2006). In the presence of a scavenge agent on superoxide radicals, there is a decreasing intensity of color generation. As shown in Table 2, the sulfated galactan F1.0 fraction has moderate scavenging activity on superoxide radicals, showing 15.39% scavenging activity at 5 mg mL−1 with EC50 of 19.61 mg mL−1.
The reducing power presented by a compound may serve as an indicator of its antioxidant potential (Sun et al. 2009). In the presence of an antioxidant agent there is reduction of K3Fe(CN)6 potassium ferrocyanide, which reacts with Fe+3 forming Prussian blue (Zou et al. 2008). This leads to a change in test solution color from yellow to blue in the presence of antioxidants; therefore, the greater the reducing activity of a compound, the greater the absorbance at 700 nm. Figure 3 shows that sulfated galactan F1.0 reducing power is dose dependent. By the studied method, in the presence of sulfated galactan F1.0 at 5 mg mL−1, the absorbance at 700 nm is 0.145. In previous studies in our laboratory with the same methodology, BHT at 4 mg mL−1 displayed 0.790 absorbance at the same wavelength. Using the equation line, we suggest that the concentration of F1.0 necessary to achieve 0.790 absorbance is 33.4 mg mL−1.

Reducing power of different concentrations of F1.0 from H. musciformis. Values are mean ± SEM (n = 3)
Lipid peroxidation is a consequence of the chain reaction caused by a reactive oxygen species leading to the generation of products such as lipid hydroperoxide, which has unpaired electrons or shows the ability to attract electrons from other molecules causing direct or indirect DNA damage (Zhu et al. 2004). The occurrence of the oxidation of unsaturated fatty acids in biological membranes promotes lipid radical regeneration and thus the destruction of lipid membranes (Zubia et al. 2007). The breakdown of these unsaturated fatty acids leads to the formation of malondialdehyde, which acts as an index to determine the extent of lipid peroxidation (Zhang et al. 2003). By lipid peroxidation, the system induced in egg yolk-sulfated galactan F1.0 promotes 57.92% inhibition of lipid peroxidation at 0.08 mg mL−1, showing the high inhibition power of sulfated galactan F1.0 in this methodology. However, it reaches a plateau and at 5 mg mL−1 displays 66.98% peroxidation inhibition with IC50 of 0.0030 mg mL−1 (Fig. 4).

Antioxidant activities of F1.0 from H. musciformis. a Lipid peroxidation inhibition; b hemolysis induced by H2O2 inhibition. Values are mean ± SEM (n = 3)
H2O2-induced hemolysis Inhibition
Erythrocytes possess an abundance of polyunsaturated fatty acids, membrane proteins, high cellular oxygen concentration, iron, and hemoglobin, and are therefore highly susceptible to oxidative damage and consequently are widely used to study oxidative damage in membranes. Erythrocyte exposure to free radicals can cause lipid peroxidation, protein damage, and hemolysis (Srour et al. 2000). Hydrogen peroxide is a reactive oxygen species presenting relatively high stability, diffusion, and involvement in signaling cascades and is thus considered an attractive oxidant model (Ugartondo et al. 2009). Figure 4 displays the protective action of sulfated galactan F1.0 under erythrocytes in the H2O2-induced hemolysis model. Sulfated galactan F1.0 at 5 mg mL−1 promotes 67.89% hemolysis inhibition with an IC50 of 0.3750 mg mL−1. These data show that sulfated galactan F1.0 possesses strong protective action.
Cytotoxic action of F1.0 on different ABO and Rh groups of red blood cells
By this methodology, F1.0 showed no cytotoxic activity on erythrocytes, which can be demonstrated by the non-occurrence of significative hemolysis in tested concentrations. No statistically significant difference was seen between erythrocytes of the Rh-positive and -negative groups or among the erythrocytes of the B, AB, and O groups. Thus, F1.0 does not cause erythrocyte membrane damage in any of the blood groups tested (Fig. 5).

Cytotoxic action of F1.0 from H. musciformis on different ABO and Rh groups of red blood cells. a Direct hemolysis on A positive and A negative erythrocytes. Erythrocytes were incubated for 1 and 6 h with different amounts of F1.0. There was no statistically significant difference between Rh groups tested (P > 0.05). b Direct hemolysis on A, B, AB, and O positive blood group erythrocytes. Erythrocytes were incubated for 1 and 6 h with 50 μg of F1.0. There was no statistically significant difference (P > 0.05) between erythrocytes of B, AB, and O blood groups. *P < 0.05; values are mean ± SEM (n = 3)
Effect of F1.0 on cell viability (MTT assay)
F1.0 showed low cytotoxic activity on 3 T3 lineage when treated for 24 and 48 h. The more pronounced cytotoxic action (Fig. 6a) was seen when this lineage was treated with 100 μg of sulfated galactan F1.0, leading to 76 ± 4.7% and 64 ± 4.8% cell viability after 24 and 48 h, respectively. After treatment for 72 h with 25, 50, and 100 μg of sulfated galactan F1.0 in HeLa lineage, the cell viability was 75.83 ± 2.51%, 72.11 ± 3.41%, and 60.73 ± 3.40%, respectively. Therefore, sulfated galactan F1.0 displays low cytotoxic action on 3 T3 cells and moderate antitumoral action on HeLa cells (Fig. 6b).

Effect of F1.0 isolated from H. musciformis on cell viability. a 3 T3 cell lineage treated with F1.0 for 24 and 48 h. b HeLa cell lineage treated with F1.0 for 72 h. **P < 0.01; ***P < 0.001. Values are mean ± SEM (n = 3)
Discussion
BHA and BHT are synthetic antioxidants (Williams et al. 1999) that are commonly used; however, they have been associated with liver damage and carcinogenesis (Grice 1988; Qi et al. 2005). Sulfated galactan fraction 1.0 (F1.0) acquired in this study was rich in polysaccharides with high sulfate content. This is contrary to the Saito and Oliveira (1990) findings that H. musciformis has a low sulfate content. However, the high sulfate content of sulfated galactan F1.0 (49.47%) is consistent with data from the previous studies of our laboratory which found lamba, iota, and kappa carrageenans that possess a high degree of sulfation (38 ± 0.06%, 27.60 ± 0.12%, and 17.90 ± 0.05%, respectively) (Souza et al. 2007) and are consistent with the De Ruiter and Rudolph (1997) studies with commercial carrageenans with high sulfate content (lamba, 38%; iota, 32%; and kappa, 22%). However, variations can occur due to differences in seaweed species. Some evidence has pointed to a relationship between the molecular weight and sulfation degree of sulfated polysaccharides from seaweeds with antioxidant activity of these polysaccharides (Zhao et al. 2004; Qi et al. 2005). This correlation was found by Tannin-Spitz et al. (2005) on polysaccharides from Porphyridium, where it was demonstrated that one of the groups of polysaccharides with the highest sulfate content (4.5%) showed 20% higher antioxidant activity than the other polysaccharide group with 3% sulfate when the antioxidant capacity was evaluated by the FOX method (Wolf 1994). Therefore, in this same study lamba carrageenan with a sulfation degree of about 40% showed a low antioxidant effect when compared with polysaccharides from Porphyridium with low sulfate content.
Sulfated galactan F1.0 from H. musciformis has high sulfate content displaying moderate or high antioxidant activity, depending on the methodology used. Thus, there is a need for more studies to clarify the relationship between sulfate content and antioxidant action.
Sulfated galactan F1.0 has high molecular weight showing two populations (one of 147 and another 155 kDa) consistent with the fact that sulfated galactan F1.0 is constituted by κ- and ι-carrageenans. Commercial carrageenans possess high molecular weight structures ranging from 100 to 1,000 kDa (Van De Velde et al. 2002). Studies of NMR by Greer et al. (1984) demonstrated that carrageenan of H. musciformis is a hybrid consisting mainly of κ-carrageenan repeating units with minor proportions of ι-carrageenan. And previously, studies of Hamilton and Carroll (1962) had shown that Hypnea extract contains a potassium sensitive fraction named κ-carrageenan. All these data support our data that Hypnea extracts contain κ-carrageenan with minor proportions of ι-carrageenan. The Zou et al. (2008) studies with sulfated polysaccharides from the lac tree (Rhus vernicifera) showed that the higher molecular weight polysaccharides display lower antioxidant activity when compared with the same polysaccharides with lower molecular weight obtained after treatment with sulfur trioxide–pyridine complex in DMSO at different temperatures and for varying periods of time. However, F1.0 obtained from H. musciformis possesses high molecular weight displaying potential antioxidant action in lipid peroxidation, H2O2-induced hemolysis inhibition, and scavenging on hydroxyl radicals.
H2O2-induced hemolysis is a process where H2O2 reacts with Fe2+ in erythrocytes leading to hydroxyl radical formation, which substantially promotes hemolysis. Zhang et al. (2003) showed sulfated polysaccharide fractions from Porphyra haitanensis with low protective action against H2O2-induced hemolysis in rat erythrocytes causing about 30% of hemolysis inhibition when the fractions were utilized at approximately 8 mg mL−1. These data show the high inhibitory action of sulfated galactan F1.0 from H. musciformis on the H2O2-induced hemolysis model in human erythrocytes. Sulfated galactan F1.0 at 0.3750 mg mL−1 reaches IC50. Besides this protective action on H2O2-induced hemolysis, sulfated galactan F1.0 does not cause significant damage action on erythrocyte membranes of different ABO and Rh blood groups.
Sulfated polysaccharides from H. musciformis show low reducing power when compared with BHT. However, sulfated galactan F1.0 at 2.5 mg mL−1, when compared with the chromatography column eluted polysaccharide F2 fraction extracted from Laminaria (Saccharina) japonica at the same concentration, shows a similar action (Wang et al. 2008).
Generation of hydroxyl radicals occurs by reaction of Fe+2 and H2O2. The sulfate group of polysaccharides can reduce hydroxyl radical generation by Fe+2 chelating activity (Zou et al. 2008). However, our results are not consistent with those of the study cited above since sulfated galactan F1.0, rich in sulfate, presents moderate antioxidant activity on hydroxyl radicals, promoting their scavenging and presenting low Fe2+ chelating activity.
Lipid peroxidation is responsible for a range of harmful events in organisms such as inflammation, cellular aging, and metabolic disorders (Wiseman and Halliwell 1996). Sun et al. (2009), evaluating the antioxidant activity of purified polysaccharides from marine fungus Penicillium sp. F23-2, showed PS1-1, PS1-2, and PS2-1 polysaccharides at 0.8 mg mL−1 displaying high inhibitory activity 42.32%, 53.58%, and 64.70%, respectively, on lipid peroxidation induced in egg yolk. In this same study, BHT at 0.8 mg mL−1 showed 81.32% peroxidation inhibition. These data reveal the strong inhibitory activity of sulfated galactan F1.0 on lipid peroxidation induced in egg yolk, where sulfated galactan F1.0 at 0.08 mg mL−1 promotes 57.92% peroxidation inhibition.
In conclusion, the sulfated galactan F1.0 from the red seaweed H. musciformis, composed mainly of polysaccharides and sulfate, possesses high antioxidant activity on some in vitro systems as well as moderate antitumoral action against HeLa cells.
References
Aruoma OI (1998) Free radicals, oxidative stress, and antioxidants in human health and disease. JAOCS 75:199–212
Athukorala Y, Kim KN, Jeon YJ (2006) Antiproliferative and antioxidant properties of an enzymatic hydrolysate from brown alga, Ecklonia cava. Food Chem Toxicol 44:1065–1074
Barahona T, Chandía NP, Encinas MV, Matsuhiro B, Zúñiga EA (2011) Antioxidant capacity of sulfated polysaccharides from seaweeds. A kinetic approach. Food Hydrocolloid 25:529–535
Belokoneva OS, Villegas E, Corzo G, Dai L, Nakajima T (2003) The hemolytic activity of six arachnid cationic peptides is affected by the phosphatidylcholine-to-sphingomyelin ratio in lipid bilayers. Biochim Biophys Acta 1617:22–30
Bixler HJ, Porse H (2011) A decade of change in the seaweed hydrocolloids industry. J Appl Phycol 23:321–335
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen Y, Xie MY, Nie SP, Li C, Wang YX (2008) Purification, composition analysis and antioxidant activity of a polysaccharide from the fruiting bodies of Ganoderma atrum. Food Chem 107:231–241
Costa LS, Fidelis GP, Cordeiro SL, Oliveira RM, Sabry DA, Câmara RBG, Nobre LTDB, Costa MSSP, Almeida-Lima J, Farias EHC, Leite EL, Rocha HAO (2010) Biological activities of sulfated polysaccharides from tropical seaweeds. Biomed Pharmacother 64:21–28
De Ruiter GA, Rudolph B (1997) Carrageenan biotechnology. Trends Food Sci Technol 8:389–395
Dietrich CP, Farias GGM, Abreu LRD, Silva LF, Leite EL, Nader HB (1995) A new approach for characterization of polysaccharides from algae: Presence of four main acidic polysaccharides in three species of the class Phaeophyceae. Plant Sci 108:143–153
Dodgson KS, Price RG (1962) A note on the determination of ester sulphate content of sulphated polysaccharides. Biochem J 84:106–110
Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F (1956) Colorimetric method for determination of sugars and related substances. Anal Chem 28:350–356
Fonseca RJC, Santos GRC, Mourão PAS (2009) Effects of polysaccharides enriched in 2,4-disulfated fucose units on coagulation, thrombosis, and bleeding. Practical and conceptual implications. Thromb Haemost 102:829–836
Formanek Z, Kerry JP, Higgins FM, Buckley DJ, Morrissey PA, Farkas J (2001) Addition of synthetic and natural antioxidants to α-tocopheryl acetate supplemented beef patties: effects of antioxidants and packaging on lipid oxidation. Meat Sci 58:337–341
Greer CW, Shomer I, Goldstein ME, Yaphe W (1984) Analysis of carrageenan from Hypnea musciformis by using κ- and ι-carrageenanases and 13C-n.m.r. spectroscopy. Carbohydr Res 129(C):189–196
Grice HC (1988) Safety evaluation of butylated hydroxyanisole from the perspective of effects on forestomach and oesophageal squamous epithelium. Food Chem Toxicol 26:717–723
Hamilton RD, Carroll JJ (1962) A Comparison of polysaccharides of Hypnea musciformis and Chondrus crispus. Nature 196:1200–1201
Hu T, Liu D, Chen Y, Wu J, Wang S (2010) Antioxidant activity of sulfated polysaccharide fractions extracted from Undaria pinnitafida in vitro. Int J Biol Macromol 46:193–198
Ke C, Qiao D, Gan D, Sun Y, Ye H, Zeng X (2009) Antioxidant activity in vitro and in vivo of the capsule polysaccharides from Streptococcus equi subsp. zooepidemicus. Carbohydr Polym 75:677–682
Knutsen SH, Myslabodski DE, Larsen B, Usov AI (1994) A modified system of nomenclature for red algal galactans. Bot Mar 37:163–169
Lahaye M (2001) Developments on gelling algal galactans, their structure and physico-chemistry. J Appl Phycol 13:173–184
Lee JB, Ohta Y, Hayashi K, Hayashi T (2010) Immunostimulating effects of a sulfated galactan from Codium fragile. Carbohydr Res 345:1452–1454
Lins KOAL, Bezerra DP, Alves APNN, Alencar NMN, Lima MW, Torres VM, Farias WRL, Pessoa C, Moraes MO, Costa-Lotufo LV (2009) Antitumor properties of a sulfated polysaccharide from the red seaweed Champia feldmannii (Diaz-Pifferer). J Appl Toxicol 29:20–26
Liu F, Ng TB (2000) Antioxidative and free radical scavenging activities of selected medicinal herbs. Life Sci 66:725–735
Liu F, Ooi VEC, Chang ST (1997) Free radical scavenging activities of polysaccharide extracts. Life Sci 60:763–771
Mao W, Li H, Li Y, Zhang H, Qi X, Sun H, Chen Y, Guo S (2009) Chemical characteristic and anticoagulant activity of the sulfated polysaccharide isolated from Monostroma latissimum (Chlorophyta). Int J Biol Macromol 44:70–74
Matsuhiro B, Conte AF, Damonte EB, Kolender AA, Matulewicz MC, Mejías EG, Pujol CA, Zúninga EA (2005) Structural analysis and antiviral activity of a sulfated galactan from the red seaweed Schizymenia binderi (Gigartinales, Rhodophyta). Carbohydr Res 340:2392–2402
Matsukawa R, Dubinsky Z, Kishimoto E, Masak K, Masuda Y, Takeuchi T, Chihara M, Yamamoto Y, Niki E, Karube I (1997) A comparison of screening methods for antioxidant activity in seaweeds. J Appl Phycol 9:29–35
Mau JL, Lin HC, Chen CC (2002) Antioxidant properties of several medicinal mushrooms. J Agric Food Chem 50:6072–6077
McHugh DJ (2003) A guide to the seaweed industry. Food & Agriculture Organisation of the United Nations (FAO) Fisheries Technical Paper. FAO, Rome
Medeiros VP, Queiroz KCS, Cardoso ML, Monteiro GRG, Oliveira FW, Chavante SF, Guimaraes LA, Rocha HAO, Leite EL (2008) Sulfated galactofucan from Lobophora variegata: anticoagulant and anti-inflammatory properties. Biochem Mosc 73:1018–1024
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
O’Sullivan L, Murphy B, McLoughlin P, Duggan P, Lawlor PG, Hughes H, Gardiner GE (2010) Prebiotics from marine macroalgae for human and animal health applications. Mar Drugs 8:2038–2064
Ohta Y, Lee J, Hayashi K, Hayashi T (2009) Isolation of sulfated galactan from Codium fragile and its antiviral effect. Biol Pharm Bull 32:892–898
Oliveira EC (1998) The seaweeds resources of Brazil. In: Critchley AT, Ohno M (eds) Seaweeds resources of the world. Japan International Cooperation Agency, Japan, pp 366–371
Percival E, McDowell RH (1967) Chemistry and enzymology of marine algal polysaccharides. Academic Press, New York, p 219
Pereira L, Amado AM, Critchley AT, Van de Velde F, Ribeiro-Claro PJA (2009) Identification of selected seaweed polysaccharides (phycocolloids) by vibrational spectroscopy (FTIR-ATR and FT-Raman). Food Hydrocolloid 23:1903–1909
Pomin VP (2009) An overview about the structure–function relationship of marine sulfated homopolysaccharides with regular chemical structures. Biopolymers 91:601–609
Qi H, Zhang Q, Zhao T, Chen R, Zhang H, Niu X, Li Z (2005) Antioxidant activity of different sulfate content derivatives of polysaccharide extracted from Ulva pertusa (Chlorophyta) in vitro. Int J Biol Macromol 37:195–199
Qi HM, Zhang QB, Zhao TT, Hu RG, Zhang K, Li Z (2006) In vitro antioxidant activity of acetylated and benzoylated derivates of polysaccharide extracted from Ulva pertusa (Chlorophyta). Bioorg Med Chem Lett 16:2441–2445
Saito RM, Oliveira EC (1990) Chemical screening of Brazilian algae producing carrageenans. Hydrobiologia 204/205:585–588
Saltarelli R, Ceccaroli P, Lotti M, Zambonelli A, Buffalini M, Casadei L, Vallorani L, Stocchi V (2009) Biochemical characterisation and antioxidant activity of mycelium of Ganoderma lucidum from Central Italy. Food Chem 116:143–151
Silva FRF, Dore CMPG, Marques CT, Nascimento MS, Benevides NMB, Rocha HAO, Chavante SF, Leite EL (2010) Anticoagulant activity, paw edema and pleurisy induced carrageenan: action of major types of commercial carrageenans. Carbohydr Polym 79:26–33
Smirnoff N, Cumbes QJ (1989) Hydroxyl radical scavenging activity of compatible solutes. Phytochemistry 28:1057–1060
Srour MA, Bilto YY, Juma M (2000) Evaluation of different methods used to measure malonyldialdehyde in human erythrocytes. Clin Hemorheol Microcirc 23:23–30
Souza MCR, Marques CT, Dore CMPG, Silva FRF, Rocha HAO, Leite EL (2007) Antioxidant activities of sulfated polysaccharides from brown and red seaweeds. J Appl Phycol 19:153–160
Sun HH, Mao WJ, Chen Y, Guo SD, Li HY, Qi XH, Chen YL, Xu J (2009) Isolation, chemical characteristics and antioxidant properties of the polysaccharides from marine fungus Penicillium sp. F23-2. Carbohydr Polym 78:117–124
Swain T, Hills WE (1959) The phenolic constituents of Prunus domestica. I. The quantitative analysis of phenolic constituents. J Sci Food Agric 10:63–68
Tannin-Spitz T, Bergman M, van-Moppes D, Grossman S, Arad SM (2005) Antioxidant activity of the polysaccharide of the red microalga Porphyridium sp. J Appl Phycol 17:215–222
Tuvikene R, Truus K, Robal M, Volobujeva O, Mellikov E, Pehk T, Kollist A, Kailas T, Vaher M (2008) The extraction, structure, and gelling properties of hybrid galactan from the red alga Furcellaria lumbricalis (Baltic Sea, Estonia). J Appl Phycol 22:51–63
Ugartondo V, Mitjans M, Torres JL, Vinardell MP (2009) Biobased epicatechin conjugates protect erythrocytes and nontumoral cell lines from H2O2-induced oxidative stress. J Agric Food Chem 57:4459–4465
Usov AI, Yarotsky SV, Shashkov AS (1980) Carbon-13 NMR spectroscopy of red algal galactans. Biopolymers 19:977–990
Usov AI, Shashkov AS (1985) Polysaccharides of algae. 34. Detection of iota-carrageenan in Phyllophora brodiaei (Turn.) J.Ag. (Rhodophyta) using 13 C-NMR spectroscopy. Bot Mar 28:367–373
Van De Velde F, Knutsen SH, Usov AL, Rollema HS, Cerezo AS (2002) 1H and 13 C high resolution NMR spectroscopy of carrageenans: application in research and industry. Trends Food Sci Technol 13:73–92
Wang J, Zhang Q, Zhang Z, Li Z (2008) Antioxidant activity of sulfated polysaccharide fractions extracted from Laminaria japonica. Int J Biol Macromol 42:127–132
Wang X, Wang J, Zhang J, Zhao B, Yao J, Wang Y (2010) Structure–antioxidant relationships of sulfated galactomannan from guar gum. Int J Biol Macromol 46:59–66
Williams GM, Iatropoulos MJ, Whysner J (1999) Safety assessment of butylated hydroxyanisole and butylated hydroxytoluene as antioxidant food additives. Food Chem Toxicol 37:1027–1038
Wiseman H, Halliwell B (1996) Damage to DNA by reactive oxygen and nitrogen species: role of inflammatory disease and progression to cancer. Biochem J 313:17–29
Wolf SP (1994) Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurment of hydroperoxides. In: Packer L (ed) Methods in enzymology. Academic Press Inc., New York, pp 182–189
Ye H, Wang K, Zhou C, Liu J, Zeng X (2008) Purification, antitumor and antioxidant activities in vitro of polysaccharides from the brown seaweed Sargassum pallidum. Food Chem 111:428–432
Yuan YV, Carrington MF, Walsh NA (2005) Extracts from dulse (Palmaria palmata) are effective antioxidants and inhibitors of cell proliferation in vitro. Food Chem Toxicol 43:1073–1081
Zhang EX, Yu LJ (1997) Studies on polysaccharide from Sargassum thumbergii for is ability to scavenge active oxygen species. Chin J Mar Drugs 3:1–4
Zhang Q, Yu P, Li Z, Zhang H, Xu Z, Li P (2003) Antioxidant activities of sulfated polysaccharide fractions from Porphyra haitanesis. J Appl Phycol 15:305–310
Zhao X, Xue CH, Li ZJ, Cai YP, Liu HY, Qi HT (2004) Antioxidant and hepatoprotective activities of low molecular weight sulfated polysaccharide from Laminaria japonica. J Appl Phycol 16:111–115
Zhu YZ, Huang SH, Tan BK, Sun J, Whiteman M, Zhu YC (2004) Antioxidants in Chinese herbal medicines: a biochemical perspective. Nat Prod Rep 21:478–489
Zou C, Du Y, Li Y, Yang J, Feng T, Zhang L, Kennedy JF (2008) Preparation of lacquer polysaccharide sulfates and their antioxidant activity in vitro. Carbohydr Polym 73:322–331
Zubia M, Robledo D, Freile-Pelegrin Y (2007) Antioxidant activities in tropical marine macroalgae from the Yucatan Peninsula, Mexico. J Appl Phycol 19:449–458
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
das Chagas Faustino Alves, M.G., Dore, C.M.P.G., Castro, A.J.G. et al. Antioxidant, cytotoxic and hemolytic effects of sulfated galactans from edible red alga Hypnea musciformis . J Appl Phycol 24, 1217–1227 (2012). https://doi.org/10.1007/s10811-011-9763-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-011-9763-3