
Abstract
The frequency, type and distribution of simple sequence repeats (SSRs) in Porphyra haitanensis genomes was investigated using expressed sequence tag (EST) data deposited in public databases. A total of 3,489 non-redundant P. haitanensis ESTs were screened for SSRs using SSRhunter software. From those, 224 SSRs in 210 ESTs were identified; trinucleotides were the most common type of SSR (64.29%), followed by dinucleotides (33.48%). Tetranucleotides, pentanucleotides, and hexanucleotides were not common. Among all identified motif types, CGG/CCG had the highest frequency (33.9%), followed by TC/AG (24.6%). From these EST-SSRs, 37 SSR primer-pairs were designed and tested using common SSR reaction conditions with 15 P. haitanensis DNAs as templates. The results showed that 28 SSR primer-pairs gave good amplification patterns. These were used to conduct SSR analyses of genetic variations of the 15 germplasm strains of P. haitanensis. A total of 224 alleles were detected, with the number of alleles ranging from 4 to 15. The effective number of alleles, expected heterozygosity, and polymorphism information content of the 15 germplasm strains of P. haitanensis were 2.81, 0.64, and 0.57, respectively. All of these parameters indicate that the 15 germplasm strains of P. haitanensis harbor rich genetic variation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The red algal genus Porphyra is an important economic marine crop, with an annual harvest of more than 130,000 t (dry weight) and a value of over $US 2 billion. Today, farming and processing of Porphyra have generated the largest seaweed industries in East Asian countries such as China, Japan, South Korea, and North Korea (Sahoo et al. 2002). Amongst Porphyra species, P. haitanensis is one of the most important. It has been cultivated widely along the coasts of South China, especially the Fujian and Zhejiang Provinces. In recent years, the production of P. haitanensis has made up 75% of the total production of cultivated Porphyra in China (Zhang et al. 2005). Although P. haitanensis seafarming has been employed since the early 1960s, most cultivated strains are wild varieties collected from the coasts. These strains have been reused over generations without selection. The work of germplasmatic purification, rejuvenation, and genetic improvements had fallen behind, resulting in the degeneration of cultivar quality. Therefore, it is important to select or breed new strains of P. haitanensis with strong economic traits and use them for cultivation. To select or breed such strains effectively, molecular genetics studies, especially the development of molecular markers, are required.
Several molecular markers have been developed for P. haitanensis for analyses of genetic diversity and germplasm identification. The methods used include the random amplification of polymorphic DNA (RAPD) (Jia et al. 2000), intersimple sequence repeats (ISSR) (Xie et al. 2007), amplified fragment length polymorphism (AFLP) (Yang et al. 2002b) and sequence-related amplified polymorphism (SRAP) (Xie et al. 2008). Microsatellites or simple sequence repeats (SSRs) markers—the most widely used molecular markers in plant molecular research—have yet to be used in P. haitanensis research.
Microsatellites or SSRs are arrays of short motifs of 1–6 base pairs in length and hyper-variable in nature. They are advantageous for use as microsatellite markers in plant genetics and molecular breeding because of their multi-allelic nature, co-dominant inheritance, ease of detection by PCR, relative abundance, extensive genome coverage, and the fact that they require only a small amount of sample DNA (Rajeev et al. 2005). During the past decade, microsatellites have proven to be the marker of choice in plant genetics and breeding research. Earlier studies on SSR marker development used primarily anonymous DNA fragments containing SSRs isolated from genomic libraries, which is time consuming and expensive. However, new sources of microsatellites based on large-scale EST sequencing projects are now being utilized. More recent studies have used computational methods to detect SSRs in the sequence data generated from EST sequencing projects. About 1– 5% of ESTs from different plant species have been found to contain SSRs suitable for marker development (Kantety et al. 2002). Specifically, EST-SSR markers have been developed for a number of plant species, including grape, rice, durum wheat, rye, barley, barrel medic, ryegrass, wheat, and cotton (Ellis and Burke 2007). As a result of the development of genomic research into Porphyra, a significant number of ESTs have been identified from cDNA libraries of gametophytes and sporophytes of several different species of Porphyra (Nikaido et al. 2000; Lee et al. 2000; Yang et al. 2002a; Xu et al. 2005; Pang et al. 2005; Fan et al. 2007). By March 2008, 5,994 ESTs from sporophytic stages of P. haitanensis were available to the public in the National Center for Biotechnology Information (NCBI) EST database, forming a foundation for EST-SSRs development in P. haitanensis.
The objectives of this research were (1) to analyze the frequency, type, and distribution of EST-SSR motifs derived from the 5,994 ESTs of P. haitanensis; (2) to develop a set of SSR markers and to provide an SSR-based analytical system in P. haitanensis; and (3) to analyze the genetic variation of 15 germplasm strains of P. haitanensis using the derived SSR markers, in order to validate these SSR markers.
Materials and methods
All 15 germplasm strains of P. haitanensis used in this study were selected and purified from the coasts of Fujian province, China, and stored in the laboratory for germplasm improvements and applications of P. haitanensis in Jimei University.
EST collection and SSR-containing EST identification
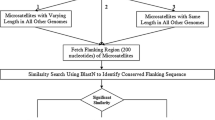
All P. haitanensis ESTs were downloaded from the dbEST database at NCBI (dbEST release 100207, http://www.ncbi.nlm.nih.gov/dbEST) and saved in FASTA format. The file was entered into a database, and redundant sequences were excluded using a combination of the programs of Clustal X (version 1.81), Treeview (version 1.61), and Genedoc (version 2.6.02). If EST sequence similarity were in excess of 90%, the ESTs would be thought of as one EST. In such cases, only the longest EST was retained, while the others were excluded. Similarly, ESTs with sequences shorter than 100 bp were also manually excluded.
The SSRhunter (version 1.61) program (Li and Wan 2005) was used to identify SSRs in the non-redundant P. haitanensisEST sequences. The parameters for the SSR search were defined as follows: the size of motifs was two to six nucleotides, and the minimum repeat unit was defined as seven for di-nucleotides, five for tri-nucleotides and four for tetra-, penta-, and hexa-nucleotides. All EST-derived SSRs were classified into three types: perfect, imperfect, and compound. Examples of perfect repeats are (AT)n or (CTG)n; imperfect repeats are (TG)n(N)x(TG)m or (GGC)n(N)x(GGC)m; and compound repeats are (GT)n(AT)m, (ATC)n(GCG)m or (CG)n(AAT)m.
Determination of SSR locations and primer design
To analyze the locations and lengths of SSRs within ESTs, the sequences were visualized using the diagram function of SSRhunter to determine if there were sufficient flanking sequences for primer design. Clones harboring an SSR at either the beginning or the end were removed, and the remaining ESTs were sent to a text file in FASTA format for primer design using Primer Premier™ 5.0. The criteria for primer design were as follows: primer length within18–24 bp; GC content between 50% and 75%; melting temperature between 50°C and 65°C; expected product size between 100 bp and 500 bp, and no secondary structure.
DNA extraction and purification
DNA was isolated from the free-living conchocelis of each germplasm strain. The collected free-living conchocelis were ground to a powder with a high-speed homogenizer, and the DNA was extracted and purified using a CTAB method (http://www.cimmyt.org/english/docs/manual/protocols/abc_amgl.pdf). DNA concentrations were determined with a DU-600 spectrophotometer (Beckman Coulter, Fullerton, CA) and adjusted to 5ng μL−1 for PCR amplification.
SSR analysis
SSR analysis was performed in a 25 μL PCR reaction mixture containing 2.5 μL 10×PCR Buffer, 5ng genomic DNA, 1.0 U Taq polymerase (Takara Biotechnology, Dalian, China), 0.2 μM forward primers (Takara), 0.2 μM reverse primer (Takara), and 200 μM dNTPs (Takara). Amplifications were performed in a MT programmable thermal controller PTC-200 (MJ Research, Waltham, MA). The following set of procedures was followed: 5 min denaturing at 94°C, 35 three-step cycles; 45 s denaturing at 94°C, 45 s annealing at given temperature (depending on the difference primer pairs) and 1 min elongation at 72°C, with a final elongation step of 10 min at 72°C. Separation of amplified fragments was accomplished by 3% agarose gel electrophoresis at 120V for 1.5 h. The amplified products were visualized under UV after staining with ethidium bromide. Microsatellite alleles were identified by their size in base pairs using Gel Works software package (version 3.0 UVP, Upland, CA).
Statistical analysis
The effective number of alleles (Ne), observed heterozygosity (Ho), and expected heterozygosity (He) were computed using PopGene software (version 3.2; Yeh et al. 1997). Polymorphism information content (PIC) was computed according to the following formula (Botstein et al. 1980): \(PIC = 1 - \sum\limits_{i = 1}^n {p_i^2 - \sum\limits_{i = 1}^{n - 1} {\sum\limits_{j = i + 1}^n {2P_i^2 P_j^2 } } } \) (P i and P j are the frequencies of the ith and jth alleles at one locus, n is the number of alleles at one locus).
Finally, corresponding cluster analysis was performed based on the Nei’s genetic distances, employing the unweighted pair-group method with arithmetic average (UPGMA) algorithm provided in the computer program NTSYSpc 2.10e (Exeter Software, Setauket, NY).
Results
Identification and characterization of EST-SSRs
After deleting redundant sequences and sequences shorter than 100 bp, a total of 3,489 non-redundant P. haitanensis ESTs (1,790 kb) were screened for SSRs using SSRhunter software. From these, 224 SSRs were identified in 210 ESTs. Mathematically, 6.02% P. haitanensis ESTs contain at least one SSR. Considering that approximately 1,790 kb was analyzed, we detected a frequency of at least one SSR per 8.0 kb in the expressed fraction of the P. haitanensis genome.
The 224 EST-SSRs contained three types of dinucleotide SSR, nine types of trinucleotide SSR, one type of tetranucleotide SSR, two types of pentanucleotide SSR, and one type of hexanucleotide SSR (Table 1). Trinucleotides were the most common type of SSR, accounting for 64.29% of P. haitanensis EST-SSRs. The second most common type of SSRs was dinucleotides, accounting for 33.48%. Tetranucleotides, pentanucleotides, and hexanucleotides were not common (Table 1).
Of the dinucleotide repeats in P. haitanensis EST-SSRs, TC/GA/CT/AG was the most common type, accounting for 73.33%. AT/TA repeats were not found, while TG/AC/CA/GT and GC/CG repeats were rare, occurring at a rate of 12% and 14.67%, respectively. Of the dinucleotide repeat types, the highest count of EST-SSRs found was 55 for TC/AG/GA/CT (Table 2).
The most common type of trinucleotide repeat was CGG/GGC/GCG/CGC/CCG/GCC, accounting for 52.78% of all trinucleotide repeats found in P. haitanensis ESTs. This was followed by AAC/CAA/ACA/GTT/TGT/TTG (18.75%) and AGC/CAG/GCA/CTG/GCT/TGC (12.5%). All other types of trinucleotide repeats were about 16%, and ACT/CTA/TAC/GTA/TAG/AGT repeats were not found. The highest number of trinucleotide repeat types found was 65 (Table 2).
To obtain a more detailed analysis of SSR structure, we used the three-class categorization proposed by Weber (1990). For the 244 EST-SSRs, we observed a proportion of 71.4% (160/224) of perfect, 28.2% (63/224) of imperfect, and 0.4% (1/224) of compound repeats (Table 2).
Designing and testing SSR primers
Not all the SSRs were suitable for primer design. Out of the 224 EST-SSRs, primer pairs could be designed for only 37. For the remaining 187 EST-SSRs, primer-pairs could not be designed for one of the following reasons: (1) SSRs were located too close to the end of the flanking region to accommodate primer design, or (2) the base composition of the flanking sequence was unsuitable. The 37 primer pairs were individually tested by carrying out SSR analyses using the 15 P. haitanensis DNAs as templates. If a primer pair gave good amplification in more than half the P. haitanensis strains tested, it was considered usable. The results indicated that 28 of the 37 designed primer pairs gave good amplification results and could be used for SSR analysis in P. haitanensis. Details of these 28 primer pairs are given in Table 3.
SSR analysis
The 15 germplasm strains of P. haitanensis were analysed with the 28 useable EST-SSR primer pairs. Figure 1 shows the SSR patterns of the P. haitanensis strains amplified by primers Phes19 and Phes28. All 28 primer pairs amplified 943 fragments, ranging from 91 bp to 486 bp in length. The amplified product of each primer pair was one locus. The numbers of alleles per locus ranged from 4 to 15 (average = 8), totaling 224 alleles.

Simple sequence repeat (SSR) patterns of the 15 germplasm strains of Porphyra haitanensis amplified by primers Phes19 and Phes28. Lanes: 1–15 Fifteen germplasm strains of P. haitanensis, M DNA marker. Arrows Positions of alleles
Analysis of the genetic variation of the 15 P. haitanensis strains (Table 4) indicated that the effective number of alleles (Ne) ranged from 1.94 to 3.79, with an average of 2.81. The expected heterozygosity (He) ranged from 0.49 to 0.75, with an average of 0.64. The polymorphism information content (PIC) was between 0.39 and 0.70, and the average was 0.57. These parameters indicated a high level of polymorphism in the 15 P. haitanensis strains, and a high degree of genetic variation.
Cluster analysis
The amplification products of the 28 EST-SSR primers pairs were used in cluster analysis of the 15 germplasm strains of P. haitanensis using the UPGMA method. Cluster analysis produced a dendrogram of these P. haitanensis strains, in which the similarity coefficient ranged from 0.32 to 0.74 (Fig. 2). The 15 germplasm strains of P. haitanensis were divided into two major groups at the 0.60 similarity level. One group comprised 12 strains and the other group comprised 3 strains.

Cluster analysis of germplasm strains of P. haitanensis (1–15) using the unweighted pair-group method with arithmetic average (UPGMA)
Discussion
Development of SSR markers for Porphyra
Simple sequence repeats have become important molecular markers for a broad range of applications. These include genome mapping and characterization, phenotype mapping, marker-assisted selection of crop plants and a range of molecular ecology and diversity studies (Ellis and Burke 2007). However, few SSRs have been used in Porphyra research because the standard methods to develop SSR markers are time-consuming and expensive. The process is extensive and requires the creation of a small-insert genomic library, subsequent hybridization with tandemly repeated oligonucleotides, and the sequencing of candidate clones. Consequently, to date, only Zuo et al. (2006) have reported 11 polymorphic SSR loci obtained from P. haitanensis through an enriched genomic library.
The development of EST sequencing projects has generated a wealth of DNA sequence information that has been incorporated into online databases (Rudd 2003). Sequence data can be downloaded from GenBank, DDBJ, and EMBL to scan for SSRs. The method of in silico screening for microsatellite markers is economically effective, thus many SSR markers have been successfully developed for several crop plants using this method (Rajeev et al. 2005). However, this approach is feasible only in species that have undergone EST sequencing projects. Recently, Porphyra yezoensis has been recognised as a model alga for basic and applied studies in marine life sciences (Sahoo et al. 2002; Waaland et al. 2004), and over 20,000 ESTs have been established. Experiments have also been performed to mine SSR markers from these ESTs. Liu et al. (2005) isolated 211 non-redundant SSR loci from 20,979 ESTs sequences of P. yezoensis, and 15 of these loci were selected for designing microsatellite primers. Sun et al. (2006) mined 391 SSRs from the 20,979 P. yezoensis ESTs with SSRIT software, and from the 391 SSRs, 48 SSR primer pairs were designed and tested under commonly used SSR reaction conditions using 22 Porphyra DNA samples as templates. The results showed that 41 SSR primer pairs gave good amplification patterns. By using bio-informatics analysis, Wang et al. (2007) discovered that 1,162 out of 21,954 ESTs of P. yezoensis contained microsatellites and 984 of these ESTs fell into 112 contigs, while the other 178 ESTs were singletons. The P. haitanensis EST project has also generated a large set of EST sequences recorded in public databases. However, to date, no SSRs had been developed from these P. haitanensis ESTs.
Characterization of EST-SSRs in P. haitanensis
SSRs are distributed in all regions of the genomic DNA of eukaryotic organisms, including both non-coding (such as introns or intergenic spaces) and coding regions (Temnykh et al. 2001). Usually, SSRs exist in 3–5% of EST sequences in land plants (Rajeev et al. 2005). In this work, 210 ESTs, which contained 224 SSRs, were identified from 3,489 non-redundant P. haitanensis ESTs (1,790 kb); approximately 6.02% of all the P. haitanensis ESTs contained SSRs. This is a higher percentage than that found in barley (3.4%), rice (4.7%), sorghum (3.6%), wheat (3.2%), maize (1.5%) (Kantety et al. 2002), and P. yezoensis (2.1%) (Sun et al. 2006). The reason for this is unclear, although it could be related to the small size of the P. haitanensis genome.
Like the statistical criterion of Cardle et al. (2000), the highest frequency of the EST-derived SSRs was found in rice, at 3.4 kb between SSRs, followed by soybean (7.4 kb), maize (8.1kb), tomato (11.1 kb), Arabidopsis (13.8 kb), poplar (14.0 kb) and cotton (20.0 kb). An overall average for these species was one SSR for every 5.4 kb (7,193 SSRs found in 38,502 kb of sequence) (Cardle et al. 2000). In P. haitanensis, the frequency of EST-derived SSRs was one SSR every 8.0 kb (224 SSRs found in 1,790 kb of sequence), which is similar to the frequency previously observed in other species.
Although criteria for screening for EST-SSRs in different plants vary, the most common SSR motifs in different plants are trinucleotide repeats (30–78%; Varshney et al. 2002; Rota et al. 2005; Wang et al. 2006). The results found in P. haitanensis are in agreement with earlier studies. Of the 224 SSRs, 144 (64.29%) were trinucleotide repeats. Among all the dinucleotide and trinucleotide repeat types in the P. haitanensis EST-SSRs, AG/TC and GGC/CCG motifs were the most common repeats, accounting for 73.33% (55/75) and 52.78% (76/144) of all dinucleotide and trinucleotide repeats, respectively. The same results have also been found in several crop plants, as described by Kantety et al. (2002) in comparative analysis using publicly available EST databases for barley, maize, rice, sorghum, and wheat.
If all four nucleotides are present in random combinations, the canonical set of SSR motifs is represented by 4 different duplets (AC, AG, AT, CG), 10 different triplets, 33 different quadruplets, 102 different quintuplet and 350 different hexad motifs (Rota et al. 2005). In the source sequences, all these basic nucleotide motifs can be represented in variant forms of the same basic set, or by their reverse complements. However, to maintain a consistency in the database for estimating frequencies, they were transformed into the canonical motifs. Reverse complements and variants would include, for example, CT for AG and GAG for AGG (Rota et al. 2005). However, in this work, most types of tetranucleotide, pentanucleotide. and hexanucleotide SSRs, one type of dinucleotide SSR (AT/TA) and one type of trinucleotide SSR (ACT/CTA/TAC/GTA/TAG/AGT) did not occur in P. haitanensis EST-SSRs. These results indicated that the distribution of different types of SSRs in P. haitanensis was obviously defective. This distribution is the actual distribution of P. haitanensis EST-SSRs. This finding requires further research, because only 3,489 EST sequences were screened in this work and thus there is insufficient evidence to explain the true nature of the distribution.
Genetic variation in 15 germplasm strains of P. haitanensis
The study of genetic variation is the basis for any breeding program. The first step in an effective breeding or conservation program is to accurately evaluate the available genetic resources. Ne, Ho, He, and PIC are all parameters of genetic variation. The value of these parameters varies with abundance (Nei 1972). SSR analysis is a well established tool for measuring genetic variation. EST-SSRs are distributed in coding regions. Furthermore, genetic variations detected by EST-SSR must accurately reflect actual gene variations.
On the basis of the present study, the variation between the 15 germplasm strains of P. haitanensis was high (Ne = 2.66, Ho = 0.64, He = 0.64 and PIC = 0.57), which could provide abundant genetic variance loci for the heredity and breeding of P. haitanensis. This result is also in agreement with earlier studies that detected genetic variation in P. haitanensis strains using other molecular markers (Jia et al. 2000; Yang et al. 2002b; Xie et al. 2008). Geographic isolation, living environment, manner of reproduction, population genetic bottleneck problems, gene flow, and selection have significant impacts on the genetic construction of populations (Hamrick 1987). In P. haitanensis, the high-level genetic variation can be attributed to the specialized life-cycle, which is determined by its progenitive manner. Primarily, the thalli of P. haitanensis are dioecious, and the carpospores and conchospores can spread as far as the ocean currents take them. The long periods of cross-fertilization and frequent gene exchange results in high-level variations.
The clustering order can reflect the relationships between strains. According to our research, the germplasm strains 13–15 and 1–12 of P. haitanensis have higher genetic identity. Melchinger et al. (1992) believed that lower genetic parental identity resulted in more obvious heterosis to a certain extent. When these germplasm strains were used in breeding, stronger heterosis was achieved from parental combinations with lower genetic identity, because there were more variations.
Abbreviations
- SSRs:
-
Simple sequence repeats
- EST:
-
Expressed sequence tags
- Ne :
-
Effective number of alleles
- He :
-
Expected heterozygosity
- Ho :
-
Observed heterozygosity
- PIC :
-
Polymorphism information contents
- UPGMA:
-
Unweighted pair-group method with arithmetic average
References
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Cardle L, Ratnsay L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Ellis JR, Burke JM (2007) EST-SSRs as a resource for population genetic analyses. Heredity 99:125–132 doi:10.1038/sj.hdy.6801001
Fan XL, Fang YJ, Hu SN, Wang GC (2007) Generation and analysis of 5318 expressed sequence tags from the filamentous sporophyte of Porphyra Haitanensis (Rhodophyta). J Phycol 43:1287–1294 doi:10.1111/j.1529-8817.2007.00415.x
Hamrick JL (1987) Gene flow and the distribution of genetic variation in plant populations. In: Urbanska KM (ed) Differentiation patterns in higher plants. Academic, New York, pp 53–67
Jia JH, Wang P, Jin DM, Qu XP, Wang Q, Li CY et al (2000) The application of RAPD Markers in diversity detection and variety identification of Porphyra. Acta Bot Sin 42:403–407
Kantety RV, La RM, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 48:501–510 doi:10.1023/A:1014875206165
Lee EK, Seo SB, Kim TH, Sung SK, An G, Lee CH et al (2000) Analysis of expressed sequence tags of Porphyra yezoensis. Mol Cell 10:338–342
Li Q, Wan JM (2005) SSRhunter: developmet of a local searching software for SSR sites. Hereditas 27:808–810
Liu BQ, Zeng QG, Luo QJ, Wang YJ, Li SH (2005) Isolation of microsafellite loci from dbEST of algae Porphyra yezoensis and primer amplification of interspecies transfer. Oceanol Limnol Sin 36:248–254
Melchinger AE, Boppenmaie J, Dhillon BS, Pollmer WG, Herrmann RG (1992) Genetic diversity for RFLPs in European maize inbreds. II Relation to performance of hybrids within versus between heterotic groups for forage traits. Theor App Genet 84:672–681
Nei M (1972) Genetic distance between populations. Anim Nat 106:283–292
Nikaido I, Asamizu E, Nakajima M, Nakamura Y, Saga N, Tabata S (2000) Generation of 10,154 expressed sequence tags from a leafy gametophyte of a marine red alga, Porphyra yezoensis. DNA Res 7:223–227 doi:10.1093/dnares/7.3.223
Pang GX, Wang GC, Hu SN, Zeng CK (2005) Acquirement and analysis of expressed sequence tags of filamentous sporophyte of Porphyra haitanensis. Oceanol Limnol Sin 36:452–458
Rota LM, Kantety RV, Yu JK, Sorrells ME (2005) Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat,and barley. BMC Genomics 6:23–34 doi:10.1186/1471-2164-6-23
Rajeev KV, Andreas G, Mark ES (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55 doi:10.1016/j.tibtech.2004.11.005
Rudd S (2003) Expressed sequence tags: alternative or complement to whole genome sequences? Trends Plant Sci 8:321–329 doi:10.1016/S1360-1385(03)00131-6
Sahoo D, Tang XR, Yarish C (2002) Porphyra—the economic seaweed as a new experimental system. Curr Sci 83:1313–1316
Sun JW, Liu T, Guo BT, Jin DM, Weng ML, Feng YB et al (2006) Development of SSR primers from EST sequences and their application in germplasm identification of Porphyra lines (Rhodophyta). Eur J Phycol 41:329–336 doi:10.1080/09670260600740906
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch SR (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452 doi:10.1101/gr.184001
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis of frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7:537–546
Waaland JR, Stiller JW, Cheng PD (2004) Macroalgae candidates for genomics. J Phycol 45:26–33
Wang CB, Guo WZ, Cai CP, Zhang TZ (2006) Characterization, development and exploitation of EST-derived microsatellites in Gossypium raimondii Ulbrich. Chin Sci Bull 51(5):557–561 doi:10.1007/s11434-006-0557-y
Wang MQ, Hu JJ, Zhuang YY, Zhang L, Liu W, Mao YX (2007) In silico screening for microsatellite markers from expressed sequence tags of Porphyra yezoensis (Bangiales, Rhodophyta). J Ocean Univ China (Oceanic and Coastal Sea Research) 6:161–166 doi:10.1007/s11802-007-0161-z
Weber JL (1990) Informativeness of human (dC-dA)n·(dG-dT)n polymorphisms. Genomics 7:524–530 doi:10.1016/0888-7543(90)90195-Z
Xie CT, Ji DH, Chen CS, Xu Y, Zhang Y (2007) Application of ISSR markers in germplasm identification of different color’s Porphyra haitanensis filament strains. J Fish Sci China 31:105–111
Xie CT, Chen CS, Ji DH, Xu Y, Xu YJ (2008) Establishment of SRAP molecular marker reaction system for Porphyra haitanensis and genetic analyses of germplasm materials. Prog Nat Sci 18:247–253
Xu MJ, Mao YX, Zhang XC, Zhou XJ, Sui ZH, Zhou HL et al (2005) Bioinformatic analysis of expressed sequence tags from sporophyte of Porphyra yezoensis. Prog Nat Sci 12:24–34
Yang GP, Shen HS, Xu P, Zhang XC (2002a) Analysis of expressed sequence tags of filamentous sporophyte of Porphyra yezoensis. High Tech Lett 12:93–97
Yang R, Liu BQ, Luo QJ, Wang YJ, Zhou XC, Dai JX (2002b) Genetic variation of Porphyra haitanensis by applying AFLP. High Tech Lett 12:83–86
Yeh FC, Young RC, Boyle TBJ, Ye ZH, Mao JX (1997) POPGENE,the user-friendly shareware for population genetic analysis. MolecularBiology and Biotechnology Centre, University of Alberta, Edmonton,Alberta, Canada
Zhang XC, Qin S, Ma JH, Xu P (2005) The genetics of marine algae. China Agriculture Press, Beijing
Zuo ZH, Wang CG, Cao XH, Su Y, Liao LJ, Chen YX (2006) Isolation and characterization of microsatellite loci from a commercial cultivar of Porphyra haitanensis. Mol Ecol Notes 7:522–524 doi:10.1111/j.1471-8286.2006.01642.x
Acknowledgments
This research was supported in part by the National High Technology Research & Development Program of China (“863” Program) (Grant No.2006AA10A413), the National Natural Science Foundation of China (Grant No.40676077), the National Natural Science Foundation of Fujian Province, China (Grant No. 2007J0064), the Construction Program of Science and Technological Platform of Fujian Province, China (Grant No. 2007N2011), and the Foundation for Young Professors of Jimei University, China (Grant No. 2007C002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xie, CT., Chen, CS., Ji, DH. et al. Characterization, development and exploitation of EST-derived microsatellites in Porphyra haitanensis Chang et Zheng (Bangiales, Rhodophyta). J Appl Phycol 21, 367–374 (2009). https://doi.org/10.1007/s10811-008-9379-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-008-9379-4