
Abstract
Efficacy and safety of 2 risperidone doses were evaluated in children and adolescents with autism. Patients (N = 96; 5–17 years), received risperidone (low-dose: 0.125 mg/day [20 to <45 kg], 0.175 mg/day [>45 kg] or high-dose: 1.25 mg/day [20 to <45 kg], 1.75 mg/day [>45 kg]) or placebo. Mean baseline (range 27–29) to endpoint change in Aberrant Behavior Checklist-Irritability (primary endpoint) was significantly greater in the high-dose—(−12.4 [6.5]; p < 0.001), but not low-dose (−7.4 [8.1]; p = 0.164) group, versus placebo (−3.5 [10.7]). Clinical Global Impressions-Severity and Children’s Yale-Brown Obsessive Compulsive Scale scores improved significantly only in the high-dose group, consistent with ABC-I results. Somnolence, sedation and increased appetite occurred more frequently in high-versus low-dose groups. Overall, increased appetite occurred most frequently.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autistic disorder is a neurological and developmental disorder that develops early in childhood and usually persists throughout life. It manifests as a deficit in reciprocal social interaction, and verbal and nonverbal communication, along with restricted or stereotyped behaviors (Schultz 2005; Libbey et al. 2005).
Recent studies have suggested some probable contributing factors for the occurrence of autism in children, including genetic predisposition (Freitag 2007), exposure to possible environmental risk factors (Szpir 2006; Persico and Bourgeron 2006) and early fetal brain injury (Limperopoulos et al. 2007).
Risperidone is a second generation antipsychotic (SGA) approved in the United States for the treatment of irritability in children and adolescents with autistic disorder (Risperdal® prescribing information, revised version 2012). Risperidone was flexibly dosed (0.5–3.5 mg/day) in all of the studies in children and adolescents (5–16 years of age) that established the efficacy of risperidone in the treatment of irritability associated with autistic disorder (Nicolson et al. 1998; McCracken et al. 2002; Malone et al. 2002; Mc Dougle et al. 2003; RUPP 2005; Troost et al. 2005; Arnold et al. 2003; Shea et al. 2004; Gagliano et al. 2004; Pandina et al. 2007). The literature cited here captures the major studies of risperidone in autism; some smaller studies are not included. Long-term safety and tolerability in autism is a major concern associated with SGAs. Given the need for long-term use of these drugs, it is essential to determine the lowest effective dose in order to minimize the potential for treatment-related adverse events, such as metabolic adverse events (including weight gain, dyslipidemia and hyperglycemia), as well as sedation and tardive dyskinesia. The initial confirmatory studies of risperidone for the treatment of autism did not effectively establish the lowest effective dose (McCracken et al. 2002; Shea et al. 2004); therefore, the current study was designed to explore a lower dose range.
This was the first study designed to evaluate the efficacy and safety of risperidone at a dose lower (based on US Food and Drug Administration recommendation) than the minimum currently recommended dose (target dose, 1 mg/day [for patients ≥20 kg]; effective dose range, 0.5–3 mg/day) for the treatment of irritability associated with autistic disorder in children and adolescents aged 5–16 years weighing ≥20 kg (Risperdal® prescribing information, revised version 2012). In addition, the study assessed the effect of risperidone in children on a number of metabolic variables of interest, including insulin resistance and glucose metabolism, and the growth hormone axis.
Methods
Study Population
Patients were screened at an initial screening appointment according to the inclusion and exclusion criteria. Several efficacy and safety assessments (e.g., electrocardiograms, laboratory assessments, extrapyramidal symptoms (EPS) scales, etc) were also performed. Up to 3 weeks were allowed to obtain the results of all assessments and to permit tapering or discontinuation of prohibited medications before the baseline assessment.
Children and adolescents enrolled in this study were of either sex, aged 5 to 17 years (inclusive), weighing at least 20 kg, with a diagnosis of autistic disorder (using Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition [DSM-IV] criteria). The diagnosis was corroborated by standard cut-off scores on the Autism Diagnostic Interview-Revised; patients had parent-rated scores of at least 18 for Aberrant Behavior Checklist-Irritability (ABC-I) subscale of the ABC-community (a revision of the ABC that eliminates references to residential placement and focuses on the community setting), hereafter referred to as “ABC” in this report (Aman et al. 1985a, b; Aman and Singh 1994). All patients had scores of at least 4 (moderately ill) for Clinical Global Impressions-Severity (CGI-S) scale (Guy 1976a), at screening and baseline. Patients were also required to have a mental age of more than 18 months, documented at any time before or at screening (measured and documented by Leiter International Performance Test—Revised or another standardized, individualized, test of intelligence).
Additionally, patients with a history of seizures were required to be seizure-free for at least 6 consecutive months or on a stable dosage of antiepileptic drugs for at least 4 weeks before screening. All patients were also prohibited from taking psychotropic medications for at least 1 week (4 weeks for fluoxetine, and 8 weeks for depot medications) before baseline. Patients were also to have normal fasting glucose and creatinine, and liver function test levels less than 1.5 times the upper limit of normal.
Exclusion criteria were a previous or current DSM-IV diagnosis of psychotic disorder or Pervasive Developmental Disorders other than autism, determined by history and clinical interview. Neurologic disorders, moderate or severe extrapyramidal symptoms or tardive dyskinesia, and lack of response to risperidone treatment in the past were also exclusionary. Finally, girls who were pregnant or breast feeding were excluded.
Patients were recruited from the investigators’ practices; however, recruitment from outside their practices was also allowed.
The Independent Ethics Committee or Institutional Review Board at each study site approved the protocol, and the study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki and in accordance with ICH Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the respective protocol. All parents (or their legally authorized representatives) of the children and adolescents fully understood the potential risks/benefits with the protocol and provided written informed consent.
Study Design
This was a randomized, 6-week, double-blind, placebo-controlled, fixed-dose, multicenter study (16 sites [consolidated to 9 pooled centers for analysis], composed of both clinical and investigative settings) conducted in the United States between 3 December, 2007 and 9 March, 2010. The study consisted of a 3-week screening phase, a 6-week randomized double-blind phase (day 1 to day 42), and a 6-month open-label extension. The details of the 6-month open-label extension will be published separately.
During the double-blind phase, eligible patients were randomized (1:1:1 ratio) to one of two fixed-dose, weight-based, regimens of risperidone or to placebo. The randomization was conducted by using randomly permuted blocks, and was stratified by center and baseline weight (20 to <45 kg or ≥45 kg). To maintain blinding, the study drugs supplied were identical in appearance and packed in identical child-resistant containers. Patients, parents or primary caregivers, and the site personnel were all blinded to treatment assignment.
Study Medication
Risperidone and its matching placebo were provided as oral solutions in glass bottles containing 100 mL of solution. Two oral solutions, one containing 0.1 mg/mL and one containing 1.0 mg/mL of risperidone were used.
Risperidone was administered at two dose levels. The low dose 0.125 mg/day (lower than those currently recommended) was selected based on US Food and Drug Administration recommendation. The high dose 1.25 mg/day was selected based on prior studies (McCracken et al. 2002; Shea et al. 2004). As the efficacious dose would vary as per the patient’s weight, for each dose levels, two dose regimens were selected as per 2 weight classes (20 to <45 kg and ≥45 kg). Assuming that the average weight of the patients in the higher weight group will be approximately 1.4 times higher than that of the patients in the lower weight group, the 2 fixed doses in the higher weight class were to be approximately 1.4 times those of the lower weight class.
For the low-dose group, patients weighing 20 to <45 kg received 0.125 mg/day, and those weighing ≥45 kg received 0.175 mg/day. For the high-dose group, patients weighing 20 to <45 kg received 1.25 mg/day and those weighing ≥45 kg received 1.75 mg/day. In both groups, risperidone was titrated up from day 1 to day 4 (initial dose was given on days 1 to 3 and increased on day 4) according to the following: in the low-dose group, from 0.05 to 0.125 mg (20 to <45 kg) and 0.075 to 0.175 mg (≥45 kg); and in the high-dose group, from 0.5 to 1.25 mg (20 to <45 kg) and 0.75–1.75 mg (≥45 kg). Study medication was administered once daily in the morning (or evening, if sedation occurred).
Concomitant Medication
Anticholinergics and antihistamines for the treatment of emergent EPS were restricted to the lowest dose and for the shortest duration possible. Similarly, hypnotic or sedative medications (lorazepam, 0.25–2 mg; or diphenhydramine up to 50 mg) were allowed if the patient had been stable on a particular dose for at least 30 days before study start.
Efficacy Assessments
The primary efficacy parameter was the mean change from baseline to double-blind endpoint (last postbaseline assessment) in the ABC-I subscale score from the ABC. The ABC is used to evaluate treatment response for aberrant behavior in patients with autism and consists of 5 subscales: Irritability, Social Withdrawal, Stereotypic Behavior, Hyperactivity/Noncompliance, and Inappropriate Speech. In all, 58 items are rated across these subscales: each scored as 0 = no problem, 1 = slight problem, 2 = moderate problem, or 3 = severe problem.
Secondary efficacy parameters included: change from baseline to endpoint in the other ABC subscale scores (Social Withdrawal, Stereotypic Behavior, Hyperactivity/ Noncompliance, and Inappropriate Speech); change from baseline to endpoint in CGI-S score (investigator assessment of illness severity and functional impairment using a 7-point scale); and change from baseline to endpoint in the Children’s Yale-Brown Obsessive Compulsive Scale (CY-BOCS) compulsions subscale score (investigator assessment of symptom severity of obsessive-compulsive behavior, using a 5-point scale) (Scahill et al. 1997). Additionally, response rate (response was defined as at least 25 % improvement in ABC-I subscale score) and percentage of patients with Clinical Global Impression of Improvement (CGI-I) ratings of “much improved” or “very much improved” at endpoint were evaluated.
For all efficacy scales, reduction in scores reflects improvement in symptoms.
Pharmacokinetic Assessment
Two blood samples (before and at least 1 h after dosing) were collected at week 6 visit to determine the plasma concentrations of risperidone and 9-hydroxyrisperidone.
Safety Assessments
Safety assessments included evaluation of treatment-emergent adverse events (TEAEs), clinical laboratory parameters, vital sign measurements, physical examination findings, 12-lead electrocardiograms, and EPS rating scales: Simpson-Angus Rating Scale (Simpson and Angus 1970), Barnes Akathisia Rating Scale, (Barnes 1989), and Abnormal Involuntary Movement Scale (Guy 1976b). Additionally, changes from baseline to double-blind endpoint in growth hormone-related factors, prolactin and insulin resistance (homeostatic model assessment of insulin resistance [HOMA-IR] formula: glucose [mmol/L] × insulin [μU/mL]/22.5) were assessed.
Efficacy and safety evaluations were completed during the visits at baseline, day 4, and weeks 1, 2, 4 and 6, or at the time of early withdrawal.
Plasma concentrations of risperidone and 9-hydroxy-risperidone, the active metabolite, were determined at week 6 as a measure of exposure.
Statistical Methods
Thirty-one patients were randomized per group in order to have 80 % power (with a standard deviation of 8 and a 2-sided significance level of 0.05) to detect a between-group difference of 6 on the ABC-I subscale, assuming that 5 % of randomized patients would have no postbaseline ABC-I assessment.
For the primary efficacy endpoint, a prespecified step-down testing procedure was employed in which the risperidone high-dose group was compared to placebo first. If the difference was statistically significant (p < 0.05), then the risperidone low-dose group was compared with placebo.
The change from baseline in ABC-I at intermediate time points (day 4 and weeks 1–6) and at endpoint was assessed using an analysis of covariance (ANCOVA) model with dose level, baseline weight group and center as factors and baseline ABC-I subscale score as covariate.
Eleven small centers (<3 patients in either baseline weight group) were combined to form 4 pooled centers according to a plan specified before randomization disclosure. Pooled center was the center factor in all analyses. Change from baseline in other ABC subscales, CGI-S, and CY-BOCS were analyzed using a similar ANCOVA model. A Cochran–Mantel–Haenszel test controlling for center and baseline weight group was used to analyze response and CGI-I.
Efficacy analyses included all patients who had received at least 1 dose of study medication and had both baseline and postbaseline efficacy measurements (intent-to-treat [ITT] population).
All safety analyses included all patients who received at least 1 dose of the study medication. Pharmacokinetic and safety results were summarized.
Results
Patient Disposition and Characteristics
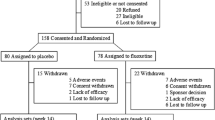
Overall, 80 % of the randomized patients (n = 77 of 96) completed the 6-week double-blind phase. Most frequent reasons for early withdrawal during the study were insufficient response (7 %) and patient choice (5 %). A higher percentage of patients in the placebo group than in either risperidone group discontinued due to insufficient response (Fig. 1).

Patient disposition. RIS risperidone. aReason not specified on case report form. bPatients were stratified based on weight into two groups, 20 kg to <45 kg and ≥45 kg
The study population was predominantly boys (88 %). Patients had a mean (SD) age of 9 (3.1) years, 77 % were <12 years of age. More than 90 % of patients were naïve to antipsychotics (Table 1). Baseline ADI-R scores were generally similar across treatment groups. The median mental age across treatment groups was 5.5 years. The median duration of treatment ranged from 42 to 43 days across the 3 groups. Antihistaminic drugs were the most commonly used concomitant medication; a higher percentage of patients in placebo (20 %; n = 7) than risperidone group (low-dose group; 7 %, n = 2 and high-dose group; 3 %, n = 1) were treated with these drugs.
Efficacy Results
Irritability scores, as measured by the ABC-I, improved significantly in the risperidone high-dose group (p < 0.001), but not in the risperidone low-dose group (p = 0.164) compared with placebo (Table 2). Separation between the risperidone high-dose and placebo groups was observed from day 8 (Fig. 2).

Least-Square Mean Change from Baseline Over Time in ABC Irritability Subscale Score (LOCF) (Intent-to-Treat Population). LOCF last observation carried forward, ABC Aberrant Behavior Checklist. A significant improvement in ABC Irritability subscale scores (p = 0.005) was observed with risperidone low dose versus placebo only on day 8. For risperidone high-dose group, the ABC Irritability subscale scores were significantly (p < 0.001) improved on all days, except day 4
Secondary Efficacy Results
Response rates were significantly higher in the risperidone high-dose group (83 %; p = 0.004), but not in the low-dose group (52 %; p = 0.817), compared with placebo (41 %). Similarly, improvements on CGI-S were significant in the high-dose-, but not in the low-dose group, compared with placebo (Table 2). The number of patients showing much or very much improvement on the CGI-I scores, was significantly higher in the risperidone high-dose group (63 %, p < 0.001), but not in the low-dose group (17 %, p = 0.985), compared with placebo (15 %).
For the ABC subscales, patients in the risperidone high-dose group showed significant improvement (p = 0.019) on the hyperactivity subscale score, and patients in the risperidone low-dose group demonstrated significant improvement on the stereotypic behavior subscale scores (p = 0.008), compared with placebo. Neither risperidone group showed significant improvement on the inappropriate speech or social withdrawal subscale scores (risperidone low-dose group p = 0.716, high-dose group p = 0.511), compared with placebo.
Consistent with the other efficacy measurements, only patients in the risperidone high-dose group showed significant improvement compared with placebo in the CY-BOCS compulsions subscale scores (risperidone high-dose group, p = 0.003; risperidone low-dose group, p = 0.454 vs. placebo).
Pharmacokinetic Results
Pharmacokinetic samples were available for 81 patients. The dose-normalized (to 1.25 mg/day), and the dose-normalized bodyweight corrected (to 0.02 mg/kg/day) plasma concentrations of active antipsychotic fraction (risperidone + 9-hydroxy-risperidone) of risperidone were similar across the low- and the high-dose risperidone group (Table 3).
Safety
Overall, the incidence of TEAEs was higher in the risperidone high-dose group (87 %) than in either the risperidone low-dose (60 %) or placebo (80 %) groups. The most common events in the combined low-and high-dose risperidone groups included: increased appetite (26 %), sedation (15 %), somnolence (11 %), and weight increase (11 %). Adverse events that occurred in at least 2 patients in the risperidone high-dose group and with twice the frequency of the risperidone low-dose group were sedation (26 vs. 3 %), somnolence (23 vs. 0 %), and increased appetite (35 vs. 17 %) (Fig. 3). Adverse events that were more common in the placebo group than either of the risperidone groups were headache (11 %), abdominal discomfort (9 %), aggression (9 %), and insomnia (6 %) (Fig. 3).

Treatment-emergent adverse events experienced by at least two patients in any treatment group (safety analysis set). Figure shows distribution of incidence rate (percentage) by adverse event for each treatment group. Symbols overlap for identical incidence rates: Risperidone low-and high-dose group; abdominal discomfort, aggression, insomnia, psychomotor hyperactivity, enuresis, headache, vomiting. Placebo group and risperidone low-dose group: ear infection, depression, epistaxis, pyrexia, thirst. Placebo group and risperidone high-dose group rash. TEAE treatment-emergent adverse event
There were no deaths in this study. Aggression was reported as a treatment-emergent serious adverse event in one patient in the placebo group, and this resulted in study discontinuation. The only other TEAE resulting in study discontinuation was sedation in one patient in the risperidone high-dose group.
The incidence of TEAEs related to somnolence (including sedation and hypersomnia) was higher in the risperidone high-dose group (55 %; n = 17) than in the risperidone low-dose group (3 %; n = 1) or the placebo group (6 %; n = 2). Extrapyramidal symptom-related adverse events during the study were most frequent in the risperidone high-dose group (16 %; n = 5) and were mostly akathisia (13 %; n = 4). No incidences of tardive dyskinesia were reported during the study.
Further, there were no clinically relevant changes from baseline in the mean AIMS total score or in BARS and SARS rating scale scores.
Mean change from baseline to double-blind endpoint in serum prolactin levels was greater in the risperidone high-dose group (20.23 ng/mL) than in the risperidone low-dose group (2.58 ng/mL) or placebo group (1.27 ng/mL). One potentially prolactin-related TEAE of oligomenorrhea was reported in one patient in the risperidone high-dose group.
Glucose metabolism-related adverse events reported were increased appetite, increased weight, and thirst (Fig. 3). The incidences of increased appetite (35 %) and increased weight (13 %) were highest in the risperidone high-dose group (Fig. 3). The change from baseline to double-blind endpoint in mean fasting glucose level was similar across all groups (−0.1 to −0.4 mg/dL; baseline values: 87–92 mg/dL). Insulin levels increased from baseline and were highest in the high-dose group from baseline to double-blind endpoint [mean [SD] change (μU/mL)]: placebo group, −0.04 [13.10]; risperidone low-dose group, 0.74 [3.09]; risperidone high-dose group, 2.36 [3.53]). Similarly, insulin resistance increased from baseline to double-blind endpoint (mean [SD] change from baseline: placebo group, −0.01 [3.05]; risperidone low-dose group, 0.14 [0.79]; risperidone high-dose group, 0.55 [0.89]). No clinically relevant change from baseline was observed in insulin growth factor-1 during the study.
Mean [SD] change from baseline to double-blind endpoint in fasting lipid values was also assessed. Triglyceride levels increased (baseline range: 65–81 mg/dL) in both risperidone low-(mean change [SD] 17.4 [40.24] mg/dL); and high-dose groups (12.4 [55.61] mg/dL), while levels decreased in the placebo group (−2.0 [28.62] mg/dL). Cholesterol levels increased (baseline values: 155–171 mg/dL) in placebo (mean change [SD] 5.6 [25.77] mg/dL) and risperidone high-dose groups (2.3 [19.37] mg/dL), and decreased (−3.8 [18.90] mg/dL) in the risperidone low-dose group. Similarly, low density lipoprotein (LDL) levels increased (baseline values: 89–97 mg/dL) in placebo (mean change [SD] 3.7 [23.34] mg/dL) and risperidone high-dose groups (0.5 [13.88] mg/dL), and decreased (−6.9 [18.37] mg/dL) in the risperidone low-dose group. High density lipoprotein (HDL) levels increased (baseline values: 54 to 56 mg/dL) in placebo (mean change [SD] 1.6 [8.85] mg/dL), but decreased in the risperidone low—(−0.2 [9.64] mg/dL) and high-dose groups (−1.9 [9.59] mg/dL).
Mean weight (SD) increased from baseline (baseline values: 36–40 kg) to double-blind endpoint for all groups: placebo, 0.7 (1.19) kg; risperidone low dose, 1.2 (1.13) kg; risperidone high dose, 2.4 (2.07) kg. Mean (SD) body mass index increased from baseline (baseline values: 19–20 kg/m2) to double-blind endpoint for all groups: placebo, 0.1 (0.67) kg/m2; risperidone low dose, 0.4 (0.70) kg/m2; and risperidone high dose, 1.1 (1.35) kg/m2.
No clinically relevant treatment-emergent changes in mean growth related factors were observed during the study. There were no clinically relevant changes in mean ECG parameters and physical examination findings.
Discussion
Long-term treatment with SGAs, such as risperidone, is associated with a variety of potentially serious adverse events such as metabolic adverse events (including weight gain, dyslipidemia and hyperglycemia), as well as sedation and tardive dyskinesia, and possible effects on growth and maturation. Hence, it is essential that children who need antipsychotic medications are treated with the lowest effective dose as they grow and mature. This study compared the effect of risperidone at doses less than the recommended dose with the currently recommended dose under controlled trial conditions in children with autistic disorder. This is the first time a placebo-controlled study has been conducted with such a low dose of risperidone and thus offers valuable information to clinicians and researchers.
Risperidone high-dose, which fell within the effective dose range of 0.5–3 mg/day (recommended in the current product label), was effective in the treatment of irritability in children and adolescents with autistic disorder. This finding is consistent with the results observed in previous studies of risperidone in children with autism (Nicolson et al. 1998; McCracken et al. 2002; Malone et al. 2002; Arnold et al. 2003). Improvement in the severity of illness and functional impairment, assessed by CGI-S, and symptoms of obsessive-compulsive behavior, assessed by CY-BOCS, was also observed in patients treated with high dose (1.25 or 1.75 mg/day) risperidone treatment. Risperidone, when used at doses less than those currently recommended (0.125 or 0.175 mg/day), did not demonstrate efficacy. Statistically significant improvement occurred only in the stereotypic behavior subscale scores.
Pharmacokinetic investigations were conducted to ensure that appreciable plasma levels were reached to aid in corroborating any noted treatment response. The systemic exposure of the active antipsychotic fraction (risperidone and 9-hydroxy risperidone) was similar in this study to those observed in a previous risperidone study in patients with autistic disorder (Aman et al. 2007; Thyssen et al. 2010).
Increased appetite, sedation, somnolence and weight increase were the most commonly reported treatment-emergent adverse events, consistent with the known profile of risperidone (Risperdal® prescribing information, revised version 2012). There were no EPS-related serious adverse events; also no incidences of tardive dyskinesia were reported.
Risperidone treatment is known to be associated with elevation of prolactin levels (Findling et al. 2003; Reyes et al. 2006; Anderson et al. 2007; Risperdal® prescribing information, revised version 2012). In the present study, increases in prolactin levels from baseline to endpoint were observed to be higher in the risperidone high-dose group, than in either the risperidone low-dose group or placebo. However, reports of potentially prolactin-related adverse events were rare (oligomenorrhea; n = 1).
Treatment–related metabolic adverse effects have always been a major concern associated with use of antipsychotics (Tschoner et al. 2007; Lieberman 2004). Most of the literature describes these effects in adult patients with schizophrenia. In the present study, risperidone did not show any clinically meaningful change in fasting glucose, cholesterol, LDL, or HDL levels from baseline to endpoint. However, the duration of the study was only 6 weeks and insulin levels and insulin resistance increased with increasing risperidone dose.
Atypical antipsychotics as a class are associated with weight gain (Turgay et al. 2002). In several trials conducted in children with autism, risperidone treatment has been associated with weight gain ranging from 2.7 to 3.7 kg (Gagliano et al. 2004; Shea et al. 2004; Malone et al. 2002; McCracken et al. 2002). In the present study, increase in mean body weight was higher in the risperidone high-dose group than in either the risperidone low-dose- or placebo groups. Psychoeducation and instituting a dietary regimen and a plan of physical activity may help in preventing or diminishing the weight gain associated with risperidone treatment (Turgay et al. 2002). Increases in mean body weight that occurred in the risperidone groups were consistent with the product labeling (Risperdal® prescribing information revised version 2012).
Any inference about the effect of risperidone on the growth hormone axis, as well as growth and sexual maturation, is limited due to the comparatively small sample size and brief study duration. Generally, safety results (including results on glucose homeostasis, metabolic parameters, and growth and sexual maturation) from the double-blind phase of this study were consistent with the known safety profile of risperidone (Risperdal® prescribing information revised version 2012) and with that of other risperidone studies in patients with autistic disorder (Nicolson et al. 1998; McCracken et al. 2002; Malone et al. 2002; Arnold et al. 2003; Aman et al. 2005).
This study has several limitations. The controlled clinical setting and specific criteria for patient enrollment may not reflect the same patients treated in other clinical settings, hence limiting the generalizability of the findings. Due to the short duration (6 weeks) of the study, the findings do not inform us about risperidone’s long-term effects in this population. Longer-term studies in this population are required to more fully assess the adverse events and metabolic changes associated with risperidone treatment. Also, studies are needed to evaluate maintenance of risperidone’s efficacy over time in children with autism.
Collectively, the data from this study demonstrate that risperidone at higher doses of 1.25 and 1.75 mg/day were efficacious; however, risperidone at doses <0.25 mg did not demonstrate significant efficacy in the treatment of irritability and related behaviors associated with autistic disorder in children and adolescents, consistent with current labeling.
References
Aman, M. G., Arnold, L. E., McDougle, C. J., Vitiello, B., Scahill, L., Davies, M., et al. (2005). Acute and long-term safety and tolerability of risperidone in children with autism. Journal of Child and Adolescent Psychopharmacology, 15(6), 869–884.
Aman, M. G., & Singh, N. N. (1994). Aberrant behavior checklist—community. (supplementary manual). East Aurora, NY: Slosson Educational Publication.
Aman, M. G., Singh, N. N., Stewart, A. W., & Field, C. J. (1985a). The Aberrant Behavior Checklist: A behavior rating scale for the assessment of treatment effects. American Journal of Mental Deficiency, 89(5), 485–491.
Aman, M. G., Singh, N. N., Stewart, A. W., & Field, C. J. (1985b). Psychometric characteristics of the Aberrant Behavior Checklist. American Journal of Mental Deficiency, 89(5), 492–502.
Aman, M. G., Vinks, A. A., Remmerie, B., Mannaert, E., Ramadan, Y., Masty, J., et al. (2007). Plasma pharmacokinetics characteristics of risperidone and their relationship to saliva concentrations in children with psychiatric or neurodevelopmental disorder. Clinical Therapeutics, 29, 1476–1486.
Anderson, G. M., Scahill, L., McCracken, J. T., McDougle, C. J., Aman, M. G., Tierney, E., et al. (2007). Effect of short- and long-term risperidone treatment on prolactin levels in children with autism. Biological Psychiatry, 61(4), 545–550.
Arnold, L. E., Vitiello, B., Mcdougle, C., Scahill, L., Shah, B., Gonzalez, N. M., et al. (2003). Parent-defined target symptoms respond to risperidone in RUPP autism study: Customer approach to clinical trials. Journal of the American Academy of Child and Adolescent Psychiatry, 42(12), 1443–1450.
Barnes, T. R. (1989). A rating scale for drug-induced akathisia. British Journal of Psychiatry, 154, 672–676.
Findling, R. L., Kusumakar, V., Daneman, D., Moshang, T., De Smedt, G., & Binder, C. (2003). Prolactin levels during long-term risperidone treatment in children and adolescents. Journal of Clinical Psychiatry, 64(11), 1362–1369.
Freitag, C. M. (2007). The genetics of autistic disorders and its clinical relevance: A review of the literature. Molecular Psychiatry, 12(1), 2–22.
Gagliano, A., Germano, E., Pustorino, G., Impallomeni, C., D’arrigo, C., Calamoneri, F., et al. (2004). Risperidone treatment of children with autistic disorder: Effectiveness, tolerability, and pharmacokinetic implications. Journal of Child and Adolescent Psychopharmacology, 14(1), 39–47.
Guy, W. (Ed.). (1976a). Clinical Global Impression (CGI). ECDEU assessment manual for psychopharmacology. National Institute of Mental Health (pp. 218–222). Rockville, Maryland: Welfare publication.
Guy, W. (Ed.). (1976b). AIMS. Abnormal Involuntary Movement Scale. ECDEU Assessment for Psychopharmacology. Rev. Ed, National Institute of Mental Health (pp. 537–543). Rockville, Maryland: Welfare publication.
Libbey, J. E., Sweeten, T. L., McMahon, W. M., & Fujinami, R. S. (2005). Review: Autistic disorder and viral infections. Journal of Neurovirology, 11, 1–10.
Lieberman, J. A. (2004). Metabolic changes associated with antipsychotic use. Prim Care Campanion. Journal of Clinical Psychiatry, 6(2), 8–13.
Limperopoulos, C., Bassan, H., Gauvreau, K., Robertson, R. L., Jr, Sullivan, N. R., Benson, C. B., et al. (2007). Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics, 120(3), 584–593.
Malone, R. P., Maislin, G., Choudhury, M. S., Gifford, C., & Delaney, M. A. (2002). Risperidone treatment in children and adolescents with autism: Short- and long-term safety and effectiveness. Journal of the American Academy of Child and Adolescent Psychiatry, 41(2), 140–147.
Mc Dougle, C. J., Stigler, K. A., & Posey, D. J. (2003). Treatment of aggression in children and adolescent with autism and conduct disorder. Journal of Clinical Psychiatry, 64(4), 16–25.
McCracken, J. T., Mcgough, J., Shah, B., Cronin, P., Hong, D., Aman, M. G., et al. (2002). Risperidone in children with autism and serious behavioral problems. New England Journal of Medicine, 347(5), 314–321.
Nicolson, R., Awad, G., & Sloman, L. (1998). An open trial of risperidone in young autistic children. Journal of the American Academy of Child and Adolescent Psychiatry, 37(4), 372–376.
Pandina, G. J., Bossie, C. A., Youssef, E., Zhu, Y., & Dunbar, F. (2007). Risperidone improves behavioral symptoms in children with autism in a randomized, double-blind, placebo-controlled trial. Journal of Autism and Developmental Disorders, 37(2), 367–373.
Persico, A. M., & Bourgeron, T. (2006). Searching for ways out of the autism maze: Genetic, epigenetic and environmental clues. Trends in Neurosciences, 29(7), 349–358.
Reyes, M., Buitelaar, J., Toren, P., Augustyns, I., & Eerdekens, M. (2006). A randomized, double-blind, placebo-controlled study of risperidone maintenance treatment in children and adolescents with disruptive behavior disorders. American Journal of Psychiatry, 163, 402–410.
Risperdal® Prescribing Information (revised 2012). http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020272s065,020588s053,021444s041lbl.pdf.
RUPP. (2005). Research units on pediatric psychopharmacology autism network. Risperidone treatment of autistic disorder: Longer-term benefits and blinded discontinuation after 6 months. American Journal of Psychiatry, 162, 1361–1369.
Scahill, L., Riddle, M. A., McSwiggin-Hardin, M., Ort, S. I., King, R. A., Goodman, W. K., et al. (1997). Children’s yale-brown obsessive compulsive scale: Reliability and validity. Journal of the American Academy of Child and Adolescent Psychiatry, 36(6), 844–852.
Schultz, R. T. (2005). Developmental deficits in social perception in autism: The role of the amygdala and fusiform face area. International Society for Developmental Neuroscience, 23, 125–141.
Shea, S., Turgay, A., Carroll, A., Schulz, M., Orlik, H., Smith, I., et al. (2004). Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics, 114(5), e634–e641.
Simpson, G. M., & Angus, J. W. (1970). A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica. Supplementum, 212, 11–19.
Szpir, M. (2006). Tracing the origins of autism: A spectrum of new studies. Environmental Health Perspectives, 114(7), A412–A418.
Thyssen, A., Vermeulen, A., Fuseau, E., Fabre, M., & Mannaert, E. (2010). Population pharmacokinetics of oral risperidone in children, adolescents and adults with psychiatric disorders. Clinical Pharmacokinetics, 49(7), 465–478.
Troost, P. W., Lahuis, B. E., Steenhuis, M. P., Ketelaars, C. E. J., Buitelaar, J. K., & Van England, H. (2005). Long-trem effects of risperidone in children with autism spectrum disorders: A placebo discontinuation study. Journal of the American Academy of Child and Adolescent Psychiatry, 44(11), 1137–1144.
Tschoner, A., Engl, J., Lamier, M., Kaser, S., Rettenbacher, M., Fleischhacker, J. R., et al. (2007). Review: Metabolic side effect of antipsychotic medication. International Journal of Clinical Practice, 61(8), 1356–1370.
Turgay, A., Binder, C., Snyder, R., & Fisman, S. (2002). Long-term safety and efficacy of risperidone for the treatment of disruptive behavior disorders in children with subaverage IQs. Pediatrics, 110(3), 1–12.
Acknowledgments
This study was funded by Johnson & Johnson Pharmaceutical Research & Development, LLC. Dr. Ananya Chikramane (SIRO Clinpharm Pvt. Ltd.) provided writing assistance and Dr. Wendy P. Battisti (Janssen Research & Development, LLC) provided additional editorial support for this manuscript. We thank Dr. Joseph Palumbo (Janssen Research & Development, LLC) for his contributions to study design and review of the manuscript, Dr. Vivek Kusamakar, in memoriam, for his significant contributions to the study, and Ms. Andrea Davis (Janssen Research & Development, LLC), the clinical trial manager for the study. The authors also thank the study participants, without whom this study would never have been accomplished and the following investigators for their participation in this study: United States: Attalla, Ashraf, M.D.; Baber, Riaz, M.D.; Bregman, Joel, M.D.; Chueh, Daniel, M.D.; Djukic, Aleksandra, M.D.; Ginsberg, Lawrence, M.D.; Greenhill, Laurence, M.D.; Hagerman, Randi, M.D.; Handal, Nelson, M.D.; Holloway, Willis, M.D.; Kolevzon, Alexander, M.D.; Lerman, Mark, M.D.; Malone, Richard P., M.D.; Marsella, Gregory, M.D.; Melmed, Raun, M.D.; Padilla, Americo, M.D.; Quintana, Humberto, M.D.; Robb, Adelaide S., M.D; Schexnayder, Donald, M.D.; Yadalam, Kashinath, M.D.
Conflict of Interest
Dr. Aman was an investigator for this study and has received research support or been a consultant for Janssen Research & Development, LLC, Bristol-Myers Squibb, Pfizer, Forest Research, and Hoffman La Roche. Drs. Ness, Singh, Hough and Kent and Mr. Karcher are employees of Janssen Research & Development, LLC. Drs. Ning and Kushner were employed by Janssen Research & Development, LLC during the design and conduct of this study. Dr. Ning is currently employed by Purdue Pharma and Dr. Kushner is at CFG Health Systems, L.L.C. All authors met ICMJE criteria and all those who fulfilled those criteria are listed as authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Registration: This phase-4 study is registered at ClinicalTrials.gov (NCT00576732).
Rights and permissions
About this article
Cite this article
Kent, J.M., Kushner, S., Ning, X. et al. Risperidone Dosing in Children and Adolescents with Autistic Disorder: A Double-Blind, Placebo-Controlled Study. J Autism Dev Disord 43, 1773–1783 (2013). https://doi.org/10.1007/s10803-012-1723-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10803-012-1723-5