
Abstract
The strongest proximal predictors of depression onset in adolescence are stressful life events (SLEs). Changes in the hypothalamic-pituitary-adrenal (HPA) axis response to stress are theorized to mediate the etiological effect of SLEs on depression onset. The goal of the current study was to examine differences in the cortisol response to a laboratory-induced stressor between youth with versus without at least one SLE in the etiologically-central 3-month period prior to depression onset. One hundred adolescents (24 first-onset depression, 18 recurrent depression, and 58 non-depressed controls) had five salivary cortisol samples collected over the course of the Trier Social Stress Test (TSST). SLEs were assessed using a rigorous contextual interview and rating system. Among those with an SLE, youth on their first onset of depression had a flatter cortisol reactivity slope relative to non-depressed adolescents, and youth on a recurrent episode of depression had a steeper recovery slope relative to first-onsets and non-depressed adolescents. In contrast, no between-group differences were found among those with no SLE prior to onset. These results suggest that differences in the HPA axis response pattern may represent a neurobiological mechanism that distinguishes depressed and non-depressed groups but only for adolescents whose depression is precipitated by SLEs. Further, this neurobiological mechanism may play a different role in the very first episode of depression than it does in recurrent episodes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Stressful life events (SLEs) are significant predictors of depression onset in adolescence (e.g., Duggal et al. 2000; Goodyer et al. 1985; Lewinsohn et al. 2001; Williamson et al. 1998). In particular, seminal studies using rigorous contextual life event interviews have demonstrated that adolescents who experience a major SLE (e.g., school expulsion; romantic relationship breakup) are three to six times more likely to develop an onset of depression in the subsequent 3 months than those who do not experience a SLE (e.g., Goodyer et al. 1985).
One neurobiological mechanism theorized to mediate the etiological effect of SLEs on depression onset is the hypothalamic-pituitary-adrenal (HPA) axis response to stress. Research has employed salivary cortisol output following a laboratory stress challenge to assess HPA axis function, as this hormone is the final output of this system. In particular, stress challenge paradigms that contain elements of social-evaluative threat and unpredictability, such as the Trier Social Stress Test (TSST; Kirschbaum et al. 1993), are especially relevant for studying stress reactivity in depression because they engage the same cortico-limbic circuitry associated with depression pathology (Cusi et al. 2012; see Dedovic et al. 2009).
Meta-analyses synthesizing research on the cortisol response to stress in depression have found mixed results. The first meta-analysis of this literature, conducted by Burke et al. (2005), found blunted cortisol reactivity and impaired cortisol recovery in response to laboratory stress challenge among depressed adults; however, neither of these differences were supported in a more recent examination of the cortisol response in depression (Ciufolini et al. 2014). Studies with depressed adolescents are fewer and also mixed, with some reporting higher cortisol reactivity (Hankin et al. 2010; Rao et al. 2008), and others finding blunted reactivity, particularly in severely depressed youth (Harkness et al. 2011). Meta-analytic studies in adolescents and adults with depression are consistent, however, in reporting marked individual variability in the temporal shape of the cortisol curve in response to stress (Burke et al. 2005; Ciufolini et al. 2014; Lopez-Duran et al. 2009). An important source of this variability in cortisol responsivity that has not been considered to date is individual differences in the extent to which depression is precipitated by SLEs.
Research on the relation of environmental stress and HPA axis function has often focused on cumulative lifetime and/or chronic stress (Alexander et al. 2009; Armbruster et al. 2011; Bosch et al. 2012; Cacioppo et al. 2000; Elzinga et al. 2008; Heim et al. 2002; Lok et al. 2012; Miller et al. 2007; Pike et al. 1997; Rao et al. 2008; van Eck et al. 1996). In general, the results of these studies suggest that a lifetime characterized by high levels of chronic stress is associated in adolescence or adulthood with higher basal cortisol and blunted cortisol reactivity to laboratory stress challenge. Specifically, chronic stress is associated with prolonged HPA axis activation, and consequent hormonal exposure, which leads to a down-regulation of the HPA axis response, particularly among individuals with depression.
Despite the important knowledge gained from the above studies regarding the effects of chronic or cumulative lifetime stress on HPA axis function, the relation of HPA axis function to SLEs that are acute, proximal, and thus most germane to depression onset, is still unknown. To our knowledge, only two studies have examined the relation of proximal and acute SLEs to HPA axis function in clinical samples. In a sample of depressed and non-depressed women, Heim et al. (2002) found no relation of negative SLEs in the past year, as assessed by self-report checklist, and peak cortisol release during the TSST in the full sample; however, they did not examine the differential relation of negative SLEs to cortisol release by depression status. Similarly, in a sample of depressed and non-depressed adolescents, Rao et al. (2008) also failed to find an association between SLEs in the past 6 months, assessed by a contextual life event interview, and peak cortisol during the TSST in the full sample; however, again, they did not examine this relation separately by depression group.
Neither of the studies with clinical samples above provided information regarding the specific relation of acute SLEs to HPA axis function in depression. Further, neither study focused on the SLEs most relevant to depression onset (i.e., those occurring 3 months prior to onset). Further, one of the above studies used a self-report checklist assessment of SLEs; such measures are highly prone to both false positive and false negative endorsements of SLEs as a function of depressive response biases (see Monroe 2008). Therefore, a strong test of the relation of etiologically-relevant SLEs to HPA axis function in depression is still needed.
A further important source of variability in depression that must be considered when examining HPA axis reactivity is the disorder’s recurrent course. As documented extensively, the relation of SLEs to the onset of depression changes as the disorder progresses from the first lifetime episode to recurrences. In particular, increasingly more minor levels of SLEs are required to trigger recurrent episodes of depression than were required to trigger its first onset (Monroe and Harkness 2005; Post 1992), a phenomenon known as stress sensitization, or kindling. One possible neurobiological mechanism that may explain kindling is progressive sensitization of the HPA axis response to stress. Indeed, individuals on a recurrent episode of depression show a more reactive cortisol response to pharmacologic challenge than those on a first episode (Gervasoni et al. 2004; Hatzinger et al. 2002). However, no studies have examined potential differences between first episode and recurrent depression in terms of the cortisol response to laboratory stress challenge tests such as the TSST. Further, no studies have tested whether the relation of SLEs prior to depression onset to HPA axis function is moderated by whether the depressive episode is a first onset or a recurrence.
The goal of the current study was to examine cortisol responses to the TSST with respect to two sources of variability in adolescents with MDD: (1) the presence versus absence of contextually defined acute SLEs in the 3-month period prior to onset; and (2) first-onset versus recurrent episode. Our sample included adolescents who met Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR; American Psychiatric Association 2000) criteria for a first or recurrent episode of a depressive disorder compared to a sample of psychiatrically healthy youth. Adolescence is a crucial developmental period for the etiology of depression; most first onsets occur in adolescence and lifetime prevalence rates grow to mirror adult levels in this developmental period (e.g., Avenevoli et al. 2015; Hankin and Abramson 1999; Kessler et al. 2001). Compared to adult-onset depression, depression that first develops in childhood or adolescence is associated with a different risk factor profile (Jaffee et al. 2002), and with a more chronic and recurrent course (Rao et al. 1995; Weissman et al. 1999; Zisook et al. 2007). Therefore, identifying neuroendocrine biomarkers associated with factors critically implicated in the etiology of depression in youth has implications for understanding the diverse endophenotypes that underlie the depression syndrome. Further, uncovering early mechanisms that drive the development of depression may lead to clear targets for intervention to curb a lifetime course of illness.
We theorize that sensitization, or kindling of the HPA axis response to stress occurs over recurrent episodes of depression via depressogenic changes in the higher order cortico-limbic circuitry that promotes heightened stress sensitivity. Further, we theorize that the kindling effects on the HPA axis will emerge over recurrence but only when primed by the occurrence of etiologically relevant SLE. Therefore, we hypothesize that depressed adolescents on a recurrent episode of depression with acute SLEs before onset will show a heightened cortisol response, as evidenced by greater reactivity and impaired recovery of the response curve, relative to those on their first onset of depression or non-depressed controls with SLEs. Further, we hypothesize that those on their first onset of depression with acute SLEs prior to onset will show a heightened cortisol response relative to non-depressed controls with SLEs in a matched time period.
It is important in any study examining the effects of acute SLEs on pathological outcomes to account for the effects of chronic stress. This is because chronic stressors can cause acute SLEs (e.g., chronic marital strife can cause an acute marital break-up event; Brown and Harris 1989) and, like SLEs, are associated with the onset of depression (Brown and Harris 1989). Further, as reviewed above, they are associated with alterations in HPA axis function. Therefore, in the current study we include chronic stress as a covariate in our models to provide a rigorous test of the hypothesis that acute and proximal SLEs are associated with differential patterns of HPA reactivity and recovery in depressed and non-depressed groups over and above the background context of chronic stress (see also Hammen et al. 2000; Harkness et al. 2006).
Method
Participants
The current study sample was collected as part of a larger study (N = 205) of stress in adolescence (Harkness et al. 2011; Stewart et al. 2013). Participants included in the current analyses were 42 depressed and 58 non-depressed youth (age range: 12–21; M = 16.09, SD = 2.37; 72 female; see Table 1 for clinical and demographic characteristics) recruited from a mid-sized community in Ontario, Canada. Depressed participants were referred by community mental health providers or recruited through advertisements. Non-depressed participants were recruited through advertisements.
All participants in the depressed group met DSM-IV-TR criteria for a current non-bipolar, non-psychotic depressive disorder of less that 2 years’ duration. The duration criterion was employed to ensure reliable recollection of the SLEs prior to onset (Brown and Harris 1978). Exclusion criteria were lifetime psychotic disorder, bipolar disorder, substance dependence, conduct disorder, developmental disability, or medical condition that could cause depression. Participants in the non-depressed group did not meet lifetime criteria for any psychiatric disorder. No female participants were pregnant at the time of the study. Some adolescents in our sample were taking antidepressant medication or oral contraceptives at the time of participation.Footnote 1
Out of the initial 205 who took part in the larger study from which we drew the current sample, 78 participants were excluded because they did not meet the inclusion or exclusion criteria for the current study. Eighteen participants were excluded for missing cortisol samples (did not complete the TSST [n = 12] or did not have enough samples to reliably estimate a cortisol curve [e.g., baseline sample only; n = 6]). A further 9 participants did not complete the SLE interview. The final sample of 100 did not differ from excluded participants on sex, age, ethnicity, or parental social status assessed using the Hollingshead index of social position (Hollingshead 1975; ps > .35).
Measures
Diagnoses and Symptoms
The child and adolescent version of the Schedule for Affective Disorders and Schizophrenia (K-SADS; Kaufman et al. 1997) was administered to assess for current and past DSM-IV diagnoses. Interviews were conducted by graduate students in clinical psychology who were trained to reliability by the senior author according to standard protocols (see Grove 1981). Interviewers met with the senior author on every case throughout the study to discuss diagnoses and interview procedure. The depressed group included 42 participants (24 first-onset, 18 recurrent) whose primary diagnoses were the following: (1) Major Depressive Disorder (n = 31); (2) Dysthymia (n = 3)Footnote 2; (3) Depressive Disorder Not Otherwise Specified (n = 6); and (4) Adjustment Disorder with Depressed Mood (n = 2).Footnote 3 The remaining 58 participants did not meet criteria for a current or past psychiatric disorder.
We administered the 21-item self-report Beck Depression Inventory-II (BDI-II; Beck et al. 1996) to assess the presence and severity of depression symptoms. Participants completed the 17-item anxious arousal (AA) subscale from the Mood and Anxiety Symptom Questionnaire (MASQ; Watson and Clark 1991) to assess symptoms of anxiety. Items were rated on a 5-point scale from 1 (not all) to 5 (extremely). The AA subscale was included in our main statistical models to control for the effect of anxiety symptoms on cortisol trajectories; however, our final results were unaffected by controlling for anxiety. Therefore, we report the simplified models below.
Pubertal Status
The Tanner Stages of Pubertal Maturation (Tanner 1962) is a physician validated (Taylor et al. 2001) assessment of pubertal development. Participants select among five illustrations of pubic hair (males and females) and breast development (females only). Higher scores (range from 1 to 5) indicate later stages of development. Pubic hair and breast development scores were averaged for female participants.
Life Events and Chronic Difficulties
The Life Events and Difficulties Schedule (LEDS-II; Bifulco et al. 1989; adolescent version, Frank et al. 1997) is a semi-structured contextual interview and rating system that assesses stressful life events and chronic stressors in several domains (e.g., health, education, relationships). Graduate student interviewers were trained to not query about the effect of stressors on the participants’ depression or their subjective reaction to identified stressors. Interviews were audiotaped and the transcribed vignettes based on interview content were rated by a team of 2–4 judges, who were unaware of participant’s depression status, using the LEDS-II manual, which contains over 5000 examples for anchoring and standardization. All interviewers and raters received extensive training and supervision in the Bedford College LEDS-II procedures by the senior author.
The contextual threat of SLEs was rated on a 5-point scale: 1 – marked, 2a – high moderate, 2b – low moderate, 3 – some, 4 – little/none (Brown and Harris 1989). Pairwise comparisons among raters ranged from κ = 0.76 to κ = 0.94. Discrepancies among raters were discussed and a consensus rating was used in analyses. For the purposes of the current analyses, we created a variable that represented the presence versus absence of a SLE of any severity occurring in the 3-month period prior to current episode onset. For non-depressed participants, we anchored events using the mean period of time between the depressed group’s onset date and the LEDS-II interview date (M = 6 months). Thus, for non-depressed participants, we included SLEs that occurred in the 3-month period that fell between 6 and 9 months before the interview date. We chose a variable representing the presence or absence of a SLE of any severity because we were interested in differentiating the cortisol trajectories of individuals whose depression was versus was not precipitated by stressful life events, including even minor stressors.Footnote 4 Further, although much of the research on the role of SLEs in the onset of depression has focused on severe events (i.e., according to convention, events rated 1-marked or 2a-high moderate) we did not have enough participants in our young sample who endorsed these events to permit analysis.
Chronic stressors were also rated by the team using the LEDS manual. The LEDS defines chronic stressors as difficulties that last at least 4 weeks, although they may last longer. We included in analyses the total number of chronic stressors of any severity present in the 3-month period prior to onset (or the matched control period). As noted above, it is important to control for the chronic context of stress in studies examining the relation of SLEs to HPA axis function because chronic stressors are associated with depression onset (Brown and Harris 1989) and with alterations in HPA axis function (Miller et al. 2007; Rao et al. 2008).Footnote 5
Cortisol Collection and Stress Task
Trier Social Stress Test (Kirschbaum et al. 1993)
A baseline cortisol sample (A) was collected after a 10 min rest period during which participants adjusted to the laboratory setting. Participants were then led into another room where two research assistants (RAs) were introduced as members of the selection committee for a human resources department. The RAs were unaware of participants’ depression status or responses to any of the questionnaire or interview material. Participants were instructed by the committee that they would be given 10 min to prepare a 5-min speech to be delivered to the committee as part of a mock job interview. They were informed that their speech would be videotaped. Participants returned to the experimental room to prepare, after which they provided a second cortisol sample (B). They then delivered the speech to the committee, and when finished, the committee surprised them with an arithmetic task that involved serially subtracting 13 s beginning at 2083. The speech and arithmetic task took approximately 15 min. Participants provided a third cortisol sample (Sample C) after completing the arithmetic task. Two additional samples were collected 40 min (D) and 80 min (E) later.
Cortisol Collection and Hormone Determinations
All participants completed the TSST between 3 and 5 pm to lessen the effect of diurnal changes in cortisol levels (Groschl 2003). Participants were instructed to refrain from eating or drinking for 1 h before their appointment. Saliva samples were collected by passive drool into 5-ml polypropylene vials (Rose Scientific Ltd, Edmonton, Alberta) and immediately stored in a freezer before being transported to a secure storage freezer (-20 C). A high sensitivity enzyme immunoassay designed for saliva (1–3002; Salimetrics LLC, State College, PA) was used to assay the resulting supernatant for cortisol. All samples for a given participant were placed on the same plate to ensure that inter-assay variability did not contribute to quantification error. Further, all samples were quantified in duplicate at 25 μl with duplicate high and low controls distributed across each plate to monitor precision. Samples with a coefficient of variation of ≥ 15 % were repeated on a different plate. The repeated samples were used to reject one of the original duplicates, and were not included in the analyses. A total of 32 assay runs were conducted in six batches. The low control, measured at 0.103 μg/dL had an intra-assay coefficient of variation of 6.2 % and an inter-assay coefficient of variation of 9.8 %. The high control, measured at 1.071 μg/dL, had an intra-assay coefficient of variation of 3.6 % and an inter-assay coefficient of variation of 5.0 %.
Procedure
Participants were scheduled for 2, 2.5-h laboratory sessions separated by about 1 week. Session 1 consisted of the diagnostic interview and questionnaires. Session 2 consisted of the TSST followed by the LEDS interview during the rest period. Participants were remunerated with $10/h. Participants in the depressed group who self-referred to the study were provided with a treatment referral.
Data Analysis
Data were analyzed using piecewise multi-level modeling (MLM) conducted with HLM 7 software. MLM offers the advantage of distinguishing within-individual change in cortisol over time from inter-individual differences in change. Consequently, MLM provides a better estimate of group-level effects and a more powerful data analytic approach than using repeated measures general linear modeling.
We set up the MLM model by nesting cortisol samples (A-E; Level 1) within participants (Level 2). To model change in cortisol across time as a function of diagnostic status (i.e., non-depressed, first-onset, or recurrent) and SLE status (i.e., present versus absent), Time (minutes) was entered as a fixed effect at Level 1, and between-subjects predictors were included at Level 2. We allowed for random intercepts and slopes to permit unique growth trajectories for each individual. We selected and report the restricted maximum likelihood method of estimating the model effects because full maximum likelihood estimates tend to have a downward bias in small samples (Snijders and Bosker 1999). Based on simulation studies of Maas and Hox (2005), our study of 100 participants was sufficiently powered to obtain unbiased estimates and standard errors for the model parameters.
The cortisol trajectory was split into two pieces, which were modeled separately, but simultaneously, as two linear components.Footnote 6 Doing so allowed us to capture the reactivity (A to C) and recovery (C to E) slopes of the cortisol trajectories in the same model. This was achieved by including two Time variables to represent each piece. This piecewise method provided us with estimates for the effect of Level 2 predictors on the intercept and slopes. Our Time variables were coded such that the intercept of our model was cortisol levels at baseline (Sample A). Slopes represented the rate of change in cortisol concentration over the reactivity and recovery periods, respectively. The Level 1 function was as follows:
where Y ti is participant i’s log cortisol value at time t, π 0i is participant i’s log cortisol value at baseline (Time variables coded as 0), π 1i and π 2i are the instantaneous rates of linear change in log cortisol for the reactivity and recovery periods, respectively, for participant i, and e ti is the residual variance in repeated measurements of log cortisol for participant i that cannot be accounted for by baseline log cortisol (π 0i) or linear change in log cortisol over time.
In our model, SLEs prior to onset were modeled dichotomously (i.e., the presence of at least one SLE before onset versus the absence of SLEs before onset). Diagnostic group was evaluated by coding the three-level variable (i.e., first-onset, recurrent, non-depressed) into two dummy coded variables. Covariates were centered at their mean. The Level 2 equations were as follows:
-
Intercept
$$ {\pi}_{0i}={\beta}_{00}+{\beta}_{01}(SLEs)+{\beta}_{02}(FirstOnset)+{\beta}_{03}(Recurrent)+{\beta}_{04}\left( Chronic\ Stress\right)+{\beta}_{05}\left( FirstOnset\ast SLEs\right)+{\beta}_{06}\left( Recurrent\ast SLEs\right)+{r}_{0i} $$ -
Reactivity Slope
$$ {\pi}_{1i}={\beta}_{10}+{\beta}_{11}(SLEs)+{\beta}_{12}(FirstOnset)+{\beta}_{13}(Recurrent)+{\beta}_{14}\left( Chronic\ Stress\right)+{\beta}_{15}\left( FirstOnset\ast SLEs\right)+{\beta}_{16}\left( Recurrent\ast SLEs\right)+{r}_{1i} $$ -
Recovery Slope
$$ {\pi}_{2i}={\beta}_{20}+{\beta}_{21}(SLEs)+{\beta}_{22}(FirstOnset)+{\beta}_{23}(Recurrent)+{\beta}_{24}\left( Chronic\ Stress\right)+{\beta}_{25}\left( FirstOnset\ast SLEs\right)+{\beta}_{26}\left( Recurrent\ast SLEs\right)+{r_2}_i $$
The inclusion of a three level factor (diagnostic status) in a regression-based model restricted us to specifying only two contrasts and did not permit examination of the main effect of diagnostic status. That is, we could compare: (1) First-Onset vs. Non-Depressed Controls; (2) Recurrent vs. Non-Depressed Controls, within any given single model, and as a function of either the presence or absence of SLEs. We report these comparisons in the results of our main statistical analyses as they best captured the differences we observed in the cortisol response curves among the three levels of the diagnostic group variable. We also examined an alternative method of specifying diagnostic group in our models that allowed for the direct comparison of First-Onsets and Recurrents. This direct comparison was not always significant on account of larger error variance among the depressed groups. Greater variance among depressed individuals is likely attributable to wider within group heterogeneity in cortisol response patterns. However, given our interest in how the First-Onsets differ from Recurrents, we also report the results of this contrast in text for comparison. Because diagnostic group was modeled by two dummy coded variables, the interaction effect in the model was represented by two variables: First-Onset X SLEs; and, Recurrent X SLEs.
Results
Preliminary Analyses
Four participants in our final sample (n = 100) were missing only one of the five cortisol samples. HLM can accommodate missing data at Level 1 by excluding the data point from analysis and assigning greater statistical weight to complete participant data; therefore we included these participants in the analyses. Prior to all analyses, a log transformation was applied to all cortisol samples to correct for the positive skew of Sample C (Skewness = 3.00). After transformation of the cortisol data, there were no statistical outliers (i.e., cases +/- 3 SD from the mean).
Those with recurrent depression were significantly older than those on their first onset and than non-depressed participants, and were more likely to be female (see Table 1). They also had a significantly longer duration of the current depressive episode and were more likely to be taking anti-depressant medication than the first-onset group. As expected, both depressed groups scored significantly higher on the BDI-II and AA than the non-depressed participants.
The first-onset (n = 16; 66.7 %), recurrent (n = 11; 61.1 %), and non-depressed (n = 40; 69.0 %) groups did not differ significantly in the number and percentage of participants with SLEs prior to onset, χ 2 (2) = 0.39, p = .823. However, the recurrent group (M = 1.94, SD = 2.15) reported significantly higher numbers of chronic stressors than the first-onset (M = 1.00, SD = 1.06) and non-depressed (M = 0.64, SD = 1.09) groups, F (2, 98) = 6.61, p = .002. Finally, there were no significant differences among the groups on any of the untransformed mean cortisol samples or in terms of pre-onset SLEs (all ps > .14).
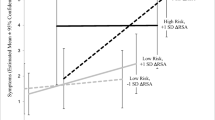
We ran our primary analyses including age, sex, and AA separately as covariates in our model. The results did not differ from those reported below; therefore, we report the model covarying only chronic stressors. Figure 1 shows the results of the estimated cortisol trajectories over time as a function of SLE and depression status based on the multilevel model including chronic stressors as a covariate. Although log transformed cortisol values were used to compute the model shown in Table 2, Fig. 1 shows the backtransformed data to facilitate cross-study comparison.

Multilevel model estimates of cortisol concentrations stratified by depression status (first-onset depression, recurrent depression, non-depressed) for those with SLEs before onset (a) and those without SLE before onset (b). Note. Cortisol concentrations were log transformed prior to analysis but backtransformed for cross study comparison
Primary Analysis
We examined the interaction of depression status (non-depressed, first-onset, and recurrent) and SLEs prior to onset (presence versus absence) on log cortisol over the course of the TSST, controlling for chronic stressors. We modeled differences in baseline cortisol (intercept at A), cortisol reactivity (slope from A to C), and cortisol recovery (slope from C to E).
We present the model estimates and simple effects in Table 2. Simple effects estimates for adolescents without SLEs are provided in brackets in-text, as are results for the First-Onset vs. Recurrent comparison.
Baseline Cortisol
As depicted in the top panel of Table 2, older age was associated with significantly higher cortisol concentration at baseline. However, neither the main effect of SLEs or its 2-way interaction with diagnostic group was significant.
Cortisol Reactivity
As depicted in the middle panel in Table 2, the main effect of SLEs on the cortisol reactivity slope approached significance (p = .058), such that the absence of SLEs was associated with a flatter (i.e., less reactive) cortisol reactivity slope than was the presence of SLEs prior to onset. This main effect was qualified by the significant 2-way interaction of First-Onset X SLEs. Among those with an SLE, participants on their first onset of depression had a flatter cortisol reactivity slope relative to non-depressed adolescents (see Table 2; the simple effect estimates for Diagnostic Status are the differences between diagnostic groups for those participants with SLEs prior to onset). However, the direct comparison of First-Onset vs. Recurrent was not significant (coefficient = 0.022, SE = 0.020, t[93] = 1.135, p = .259).
Among those without SLEs, neither the first-onset or recurrent participants differed significantly from non-depressed participants, or each other, in terms of their cortisol reactivity slope (First-Onset vs. Non-Depressed: coefficient = 0.058, SE = 0.043, t[93] = 1.344, p = .182; Recurrent vs. Non-Depressed: coefficient = 0.037, SE = 0.047, t[93] = 0.788, p = .433; First-Onset vs. Recurrent: coefficient = 0.010, SE = 0.027, t[93] = −0.391, p = .697).
Cortisol Recovery
As depicted in the bottom panel of Table 2, neither the main effect of SLEs nor the 2-way interaction effect of diagnostic group and SLEs on the cortisol recovery slope was significant. Simple effects analysis revealed that those with recurrent depression with SLEs prior to onset had a significantly steeper recovery slope compared to non-depressed participants. However, this effect should be interpreted with caution because it was detected in the absence of a significant interaction term. Steeper cortisol recovery was also found when youth with recurrent depression were compared directly to first-onset youth with SLEs prior to onset (coefficient = −0.010, SE = 0.005, t[93] = −2.552, p = .012).
Depressed first-onset and recurrent participants without SLEs before onset did not differ from non-depressed participants, or each other, in terms of their recovery slope (First-Onset vs. Non-Depressed: coefficient = 0.002, SE = 0.012, t[93] = 1.70, p = .866; Recurrent vs. Non-Depressed: coefficient = −0.002, SE = 0.013, t[93] = −0.151, p = .880; First-Onset vs. Recurrent: coefficient = −0.002, SE = 0.007, t[93] = −0.270, p = .787).
Discussion
The current study is the first to demonstrate a significant relation of stressful life events (SLEs) that occur in the most etiologically important 3-month period prior to the onset of depression and the HPA axis response to stress. The results provide novel evidence that the relation of etiologically-relevant SLEs and cortisol responsivity varies as a function of depression history. Among youth on their very first episode of depression, the presence of at least one SLE prior to onset was associated with blunted cortisol reactivity to the TSST compared to non-depressed youth. Further, among youth on a recurrent episode of depression, SLEs prior to onset were associated with steeper cortisol recovery compared to first onset and non-depressed youth. In contrast, no group differences in either cortisol reactivity or recovery were found in those without SLEs prior to onset. Importantly, the relations of acute and proximal SLEs to HPA axis function emerged above and beyond the effect of chronic stress exposure.
Research on HPA axis function in depression has shown wide heterogeneity in the cortisol response among adolescents and adults. Our results address this heterogeneity by providing evidence that individual differences in stress-related depression etiology and depression course moderate HPA axis function. As such, these results suggest that changes in HPA axis functioning may represent a neurobiological mechanism that distinguishes depressed and non-depressed groups, but only for the subset of depressed adolescents whose depression is precipitated by SLEs. Further, our results suggest that this neurobiological mechanism may play a different role in the onset of the very first episode of depression than it does in recurrent episodes.
In contrast to our hypotheses, adolescents on a recurrent episode of depression with SLEs did not show significantly greater cortisol reactivity than the other two groups. However, they did show a significantly steeper recovery slope than both the first-onset and non-depressed groups. While it is important to note that these simple effects occurred in the absence of a significant higher-order interaction, we offer the following tentative interpretation given its relevance for our a priori specified hypotheses. Specifically, we suggest that youth with recurrent depression and SLEs prior to onset had further to fall to re-regulate the HPA axis system from an already elevated baseline and/or peak. Although not statistically significant, the recurrent depressed group had a higher initial sample (Sample A) and peak (Sample C) than the first-onset and non-depressed groups, which implies greater total cortisol output in the reactivity portion of the curve. However, all three groups ended up at nearly identical levels following recovery (Sample E). We suggest that adolescents with a recurrent history of stress-induced depression may have been particularly sensitive to the laboratory environment, and thus excreted higher levels of cortisol simply in anticipation of participating in the research study. As such, their baseline (Sample A) cortisol may have been elevated relative to their biologically relevant baseline (Sample E). As a result of this potentially heightened sensitivity, and HPA axis reactivity, to the relatively minor stress of the laboratory environment among depressed adolescents on a recurrent episode, these adolescents had a steeper recovery trajectory back to equal baseline levels across groups. Thus, these steeper recovery results lend support to the presence of an intact mechanism for re-regulating HPA axis functioning from peak cortisol back to baseline following the offset of the stressor. However, our interpretation of the recovery results is based on the assumption of differences in the cortisol response trajectories even before the stress task began; thus, further examination of individual differences in acclimatization to the laboratory environment may, in and of itself, yield important information about the wide heterogeneity in cortisol response trajectories in depression (Balodis et al. 2010).
Adolescents on a first-onset of depression with SLEs, in contrast, showed a pattern of blunted cortisol reactivity in comparison with the non-depressed group, but did not differ significantly in terms of reactivity from the recurrent group. Failure to detect a statistically significant difference between the first-onset and recurrent group in the reactivity portion of the curve is likely due to greater heterogeneity in cortisol response patterns among our depressed participants than among the non-depressed group (i.e., greater error variance). One physiological process that may account for blunting of the HPA axis response to stress is down-regulation of CRH receptors on ACTH-producing cells in the anterior pituitary (Heim et al. 2000). Two factors that have been proposed to moderate this mechanism are genetic differences in glucocorticoid receptor resistance (Holsboer et al. 1995; Modell et al. 1998) and repeated or prolonged environmental stress exposure (Fries et al. 2005). Consistent with the hypothesized role of these two factors in facilitating a blunted cortisol response, cortisol blunting in response to the TSST has been reported in adolescents with severe depression and a chronic history of childhood maltreatment (Harkness et al. 2011), and samples that would be expected to have a high genetic loading, including depressed children with pre-pubertal onset (Hankin et al. 2010). Therefore, our results converge with prior studies to suggest that blunted HPA axis reactivity may represent an endophenotype of depression that is associated with an early first onset and activated by stress.
An important question that remains from our results is why stress-precipitated depression was associated with a blunted pattern of cortisol output among adolescents on the very first episode of depression, but with a more reactive pattern for those on a recurrence. On the one hand, individuals who show initial blunting of the stress response at first-onset may go on to develop heightened reactivity in subsequent episodes, through a process of sensitization to the recurrent episodes of depression. On the other hand, not all adolescents who develop a first onset will go on to have a recurrence, and, thus, our recurrent group represents only a subset of their first onset cohort that may have had, even from the beginning, a very different pattern of cortisol reactivity. In order to address this critical distinction, prospective longitudinal designs are needed to examine within-individual changes in HPA axis function from prior to depression onset through the first onset and to recurrent episodes. Nevertheless, the current results provide an intriguing first look at how the HPA axis response to stress may correspond with the role of SLEs in adolescent depression (Monroe and Harkness 2005; Post 1992).
Another important question for future research is whether our results will generalize to adult-onset depression. Meta-analytic studies that have synthesized the cortisol response patterns of depressed adults (Burke et al. 2005) yield a pattern of responsivity that greatly differs from the cortisol trajectories that we and other researchers have found in adolescent depression (Hankin et al. 2010; Lopez et al. 2009; Rao et al. 2008). Therefore, while we speculate that it is unlikely our results will directly translate to adult-onset depression, examining the relation of SLEs before onset and episode status in adults is of critical importance to further our understanding of the relative contribution of factors that affect regulation of the cortisol response to stress, as well as to understanding the precise role of the HPA axis in the etiological pathway to depression, in adolescence versus adulthood. Longitudinal studies that examine the within-subject changes in cortisol trajectories from childhood into adulthood as a function of episode status will also be important for illuminating the important developmental trajectories of HPA axis function in depression.
The results of the current study should be considered in light of the following limitations. First, we had a modest sample size to examine the interaction of SLEs and depression history status. We applied a more sophisticated data analysis strategy (i.e., multilevel modeling) than traditional general linear model techniques in order to maximize the power to detect our hypothesized effects. Nevertheless, replication in larger samples is needed. Our small sample also precluded investigation of important moderators of the above effects, such as sex, age, and anxiety or externalizing disorder comorbidity. Further, we were unable to explicitly model the interactive effects of chronic stressors and acute SLEs. Second, this was a volunteer community sample that was ethnically homogenous. Consequently, our results may not generalize to more diverse epidemiological or patient samples, or to samples in which first-onsets occur later in adulthood or earlier in childhood. Third, we did not have data on menstrual cycle phase or complete data on oral contraceptive use, although the inclusion of gender and pubertal status in our statistical analyses did not change our results. Fourth, our design was cross-sectional and thus we cannot make conclusions regarding changes in HPA axis function over the recurrent course of depression. Large-scale longitudinal designs that follow adolescents to the first onset of depression and across recurrence are required to provide a prospective test of the stress sensitization model.
Finally, the LEDS is a retrospective interview, thus raising the concern of recall bias. The LEDS helps to minimize bias in a number of ways. For example, interviewers are trained not to query respondents’ subjective perception of events, or the effect of events on respondents’ depression. Further, raters anchor their judgments using a standardized manual. The contextual approach to defining and rating SLEs has consistently shown superior reliability and validity to self-report checklist assessments of stress (see Karg et al. 2011; Monroe 2008). This contextual approach also provides rich measurement of stress exposure that we were unable to fully exploit given our limited sample size. In particular, due to low event frequencies we were unable to restrict our analyses to the sorts of severely stressful life events that have been most strongly implicated in the etiology of depression (Brown and Harris 1989), or to examine the differential relation of severe versus non-severe SLEs to HPA axis function in first-onset versus recurrent groups (Monroe and Harkness 2005). Similarly, we were unable to examine the specific relation of particular types of events (e.g., dependent versus independent; interpersonal versus non-interpersonal events) to HPA axis function. Therefore, these more fine-grained analyses await further research with larger sample sizes. Such analyses will not only provide a more complete test of the stress sensitization hypothesis, but would also permit the examination of additional stress theories in depression, such as stress generation (e.g., Hammen and Shih 2008).
Our results highlight the heterogeneity in cortisol response patterns in depression, even among those early in the course of the disorder. Specifically, in this sample of depressed youth, differences in cortisol reactivity and recovery relative to non-depressed youth were seen only when the episodes were preceded by SLEs. These SLE-related differences at the most critical developmental period for depression onset may reflect heightened sensitivity to the depressogenic effects of SLEs as adolescents and young adults learn to cope with life events more independently. Further, different portions of the cortisol trajectory were affected depending on whether the episode was a first onset or a recurrence. Specifically, we found that depressed youth with SLEs prior to onset had blunted cortisol reactivity to the TSST if they were on their first onset of depression, and a steeper cortisol recovery following the TSST if they were on a recurrence. Depression that onsets early is associated with a more chronic course; thus, these distinct neurobiological differences may also help to differentiate those at heightened risk for multiple episodes. Consequently, this avenue of research may help to inform treatment strategies aimed at targeting specific pathophysiologies by identifying neuroendocrine biomarkers of the diverse endophenotypes that comprise major depression.
Notes
We did not have complete data on oral contraceptive use for our sample; however, there were no significant differences in the distribution of users across the study variables. We ran our main analyses below controlling for the use of antidepressant medication. The pattern of our results remained unchanged. Results are available upon request.
These participants met the adolescent criteria for dysthymia, which requires only a 1-year duration.
Results of the main statistical model including only those participants who met strict criteria for MDD did not differ from the full sample. Results are available upon request.
We also ran our analyses with SLEs defined as a continuous measure of cumulative threat (i.e., the sum of the contextual threat rating for all SLEs in the relevant 3-month period). Although the pattern of results was the same, the models were not significant, suggesting that the dichotomous event definition may be most powerful in reflecting the nature of the relation between environmental triggering stress and HPA axis function.
Although some comparisons no longer reached conventional statistical levels of significance, removing chronic stress as a covariate from our models did not change the pattern of results.
Other researchers have fit the curve to a polynomial function (i.e., linear, quadratic, cubic etc.). However, we had fewer sampling points and, thus, a piecewise MLM approach provided a better fit for our data.
References
Alexander, N., Kuepper, Y., Schmitz, A., Osinsky, R., Kozyra, E., & Hennig, J. (2009). Gene-environment interactions predict cortisol responses after acute stress: implications for the etiology of depression. Psychoneuroendocrinology, 34, 1294–1303.
American Psychiatric Association. (2000). Diagnostic and statistical manual of mental disorders (4th ed., text rev.). Washington, DC: Author.
Armbruster, D., Mueller, A., Strobel, A., Lesch, K. P., Brocke, B., & Kirschbaum, C. (2011). Predicting cortisol stress responses in older individuals: influence of serotonin receptor 1A gene (HTR1A) and stressful life events. Hormones and Behavior, 60, 105–111.
Avenevoli, S., Swendsen, J., He, J. P., Burstein, M., & Merikangas, K. R. (2015). Major depression in the National Comorbidity Survey–Adolescent Supplement: prevalence, correlates, and treatment. Journal of the American Academy of Child and Adolescent Psychiatry, 54, 37–44.
Balodis, I. M., Wynne-Edwards, K. E., & Olmstead, M. C. (2010). The other side of the curve: examining the relationship between pre-stressor physiological responses and stress reactivity. Psychoneuroendocrinology, 35, 1363–1373.
Beck, A. T., Steer, R. A., & Brown, G. K. (1996). Beck depression inventory manual (2nd ed.). San Antonio: Psychological Corporation.
Bifulco, A., Brown, G., Edwards, A., Harris, T., Neilson, E., Richards, C., & Robinson, R. (1989). Life events and difficulties schedule (LEDS-2) Vol. 1: Life events manual. London: Royal Halloway and Bedford New College, University of London.
Bosch, N. M., Riese, H., Reijneveld, S. A., & Bakker, M. P. (2012). Timing matters: long term effects of adversities from prenatal period up to adolescence on adolescents’ cortisol stress response. The TRAILS study. Psychoneuroendocrinology, 37, 1439–1447.
Brown, G. W., & Harris, T. O. (1978). The Bedford College life-events and difficulty schedule: Directory of contextual threat ratings of events. London: Bedford College, University of London.
Brown, G. W., & Harris, T. O. (1989). Depression. In G. W. Brown & T. Harris (Eds.), Life Events and Illness (pp. 49–93). New York: Guilford Press.
Burke, H. M., Davis, M. C., Otte, C., & Mohr, D. C. (2005). Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology, 30, 846–856.
Cacioppo, J. T., Burleson, M. H., Poehlmann, K. M., Malarkey, W. B., Kiecolt-Glaser, J. K., Berntson, G. G., & Glaser, R. (2000). Autonomic and neuroendocrine responses to mild psychological stressors: effects of chronic stress on older women. Annals of Behavioral Medicine, 22, 140–148.
Ciufolini, S., Dazzan, P., Kempton, M. J., Pariante, C., & Mondelli, V. (2014). HPA axis response to social stress is attenuated in schizophrenia but normal in depression: evidence from a meta-analysis of existing studies. Neuroscience & Biobehavioral Reviews, 47, 359–368.
Cusi, A., Nazarov, A., Holshausen, K., MacQueen, G., & McKinnon, M. (2012). Systematic review of the neural basis of social cognition in patients with mood disorders. Journal of Psychiatry & Neuroscience, 37, 154–169.
Dedovic, K., Duchesne, A., Andrews, J., Engert, V., & Pruessner, J. C. (2009). The brain and the stress axis: the neural correlates of cortisol regulation in response to stress. NeuroImage, 47, 864–871.
Duggal, S., Malkoff-Schwartz, S., Birmaher, B., Anderson, B. P., Matty, M. K., Houk, P. R., & Frank, E. (2000). Assessment of life stress in adolescents: self-report versus interview methods. Journal of the American Academy of Child and Adolescent Psychiatry, 39, 445–452.
Elzinga, B. M., Roelofs, K., Tollenaar, M. S., Bakvis, P., van Pelt, J., & Spinhoven, P. (2008). Diminished cortisol responses to psychosocial stress associated with lifetime adverse events. Psychoneuroendocrinology, 33, 227–237.
Frank, E., Matty, M. K., & Anderson, B. (1997). Interview schedule for life-events and difficulties adolescent version (Pittsburgh). Pittsburgh: University of Pittsburgh Medical School.
Fries, E., Hesse, J., Hellhammer, J., & Hellhammer, D. H. (2005). A new view on hypocortisolism. Psychoneuroendocrinology, 30, 1010–1016.
Gervasoni, N., Bertschy, G., Osiek, C., Perret, G., Denis, R., Golaz, J., & Aubry, J. M. (2004). Cortisol responses to combined dexamethasone/CRH test in outpatients with a major depressive episode. Journal of Psychiatric Research, 38, 553–557.
Goodyer, I., Kolvin, I., & Gatzanis, S. (1985). Recent undesirable life events and psychiatric disorder in childhood and adolescence. British Journal of Psychiatry, 147, 517–523.
Groschl, M. (2003). Circadian rhythm of salivary cortisol, 17-hydroxyprogesterone, and progesterone in healthy children. Clinical Chemistry, 49, 1688–1691.
Grove, W. M. (1981). Reliability studies of psychiatric diagnosis: theory and practice. Archives of General Psychiatry, 38, 408–413.
Hammen, C., & Shih, J. H. (2008). Stress generation and depression. Risk Factors in Depression, 409–428.
Hammen, C., Henry, R., & Daley, S. E. (2000). Depression and sensitization to stressors among young women as a function of childhood adversity. Journal of Consulting and Clinical Psychology, 68, 782.
Hankin, B. L., & Abramson, L. Y. (1999). Development of gender differences in depression: description and possible explanations. Annals of Medicine, 31, 372–379.
Hankin, B. L., Badanes, L. S., Abela, J. R. Z., & Watamura, S. E. (2010). Hypothalamic-pituitary-adrenal axis dysregulation in dysphoric children and adolescents: cortisol reactivity to psychosocial stress from preschool through middle adolescence. Biological Psychiatry, 68, 484–490.
Harkness, K. L., Bruce, A. E., & Lumley, M. N. (2006). The role of childhood abuse and neglect in the sensitization to stressful life events in adolescent depression. Journal of Abnormal Psychology, 115, 730.
Harkness, K. L., Stewart, J. G., & Wynne-Edwards, K. E. (2011). Cortisol reactivity to social stress in adolescents: role of depression severity and child maltreatment. Psychoneuroendocrinology, 36, 173–181.
Hatzinger, M., Hemmeter, U. M., Baumann, K., Brand, S., & Holsboer-Trachsler, E. (2002). The combined DEX-CRH test in treatment course and long-term outcome of major depression. Journal of Psychiatric Research, 36, 287–297.
Heim, C., Ehlert, U., & Hellhammer, D. H. (2000). The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology, 25, 1–35.
Heim, C., Newport, D. J., Wagner, D., Wilcox, M. M., Miller, A. H., & Nemeroff, C. B. (2002). The role of early adverse experience and adulthood stress in the prediction of neuroendocrine stress reactivity in women: a multiple regression analysis. Depression and Anxiety, 15, 117–125.
Hollingshead, A. B. (1975). Four factor index of social status. Unpublished manuscript. Yale University, New Haven, CT.
Holsboer, F., Lauer, C. J., Schreiber, W., & Krieg, J. C. (1995). Altered hypothalamic-pituitary-adrenocortical regulation in healthy subjects at high familial risk for affective disorders. Neuroendocrinology, 62, 340–347.
Jaffee, S. R., Moffitt, T. E., Caspi, A., Fombonne, E., Poulton, R., & Martin, J. (2002). Differences in early childhood risk factors for juvenile-onset and adult-onset depression. Archives of General Psychiatry, 59, 215–222.
Karg, K., Burmeister, M., Shedden, K., & Sen, S. (2011). The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Archives of General Psychiatry, 68, 444–454.
Kaufman, J., Birmaher, B., Brent, D., Rao, U., Flynn, C., Moreci, P., & Ryan, N. (1997). Schedule for affective disorders and schizophrenia for school-age children – present and lifetime version (K-SADS-PL): initial reliability and validity data. Journal of the American Academy of Child and Adolescent Psychiatry, 36, 980–988.
Kessler, R. C., Avenevoli, S., & Merikangas, K. R. (2001). Mood disorders in children and adolescents: an epidemiologic perspective. Biological Psychiatry, 49, 1002.
Kirschbaum, C., Pirke, K. M., & Hellhammer, D. H. (1993). The ‘Trier Social Stress Test’ - A tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology, 28, 76–81.
Lewinsohn, P. M., Joiner, T. E., Jr., & Rohde, P. (2001). Evaluation of cognitive diathesis-stress models in predicting major depressive disorder in adolescents. Journal of Abnormal Psychology, 110, 203–215.
Lok, A., Mocking, R. J. T., Ruhé, H. G., Visser, I., Koeter, M. W. J., Assies, J., & Schene, A. H. (2012). Longitudinal hypothalamic-pituitary-adrenal axis trait and state effects in recurrent depression. Psychoneuroendocrinology, 37, 892–902.
Lopez-Duran, N. L., Kovacs, M., & George, C. J. (2009). Hypothalamic-pituitary-adrenal axis dysregulation in depressed children and adolescents: a meta-analysis. Psychoneuroendocrinology, 34, 1272–1283.
Maas, C. J. M., & Hox, J. J. (2005). Sufficient sample sizes for multilevel modeling. Methodology: European Journal of Research Methods for the Behavioral and Social Sciences, 1, 86–92.
Miller, G. E., Chen, E., & Zhou, E. S. (2007). If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychological Bulletin, 133, 25–45.
Modell, S., Lauer, C. J., Schreiber, W., Huber, J., Krieg, J., & Holsboer, F. (1998). Hormonal response pattern in the combined DEX-CRH test is stable over time in subjects at high familial risk for affective disorders. Neuropsychopharmacology, 18, 253–262.
Monroe, S. M. (2008). Modern approaches to conceptualizing and measuring human life stress. Annual Review of Clinical Psychology, 4, 33–52.
Monroe, S. M., & Harkness, K. L. (2005). Life stress, the kindling hypothesis, and the recurrence of depression: considerations from a life stress perspective. Psychological Review, 112, 417–445.
Pike, J. L. J., Smith, T. L. T., Hauger, R. L. R., Nicassio, P. M. P., Patterson, T. L. T., McClintick, J. J., & Irwin, M. R. M. (1997). Chronic life stress alters sympathetic, neuroendocrine, and immune responsivity to an acute psychological stressor in humans. Psychosomatic Medicine, 59, 447–457.
Post, R. M. (1992). Transduction of psychosocial stress into the neurobiology of recurrent affective disorder. American Journal of Psychiatry, 149, 999–1010.
Rao, U., Ryan, N. D., Birmaher, B., Dahl, R. E., Williamson, D. E., Kaufman, J., & Nelson, B. (1995). Unipolar depression in adolescents: clinical outcome in adulthood. Journal of the American Academy of Child and Adolescent Psychiatry, 34, 566–578.
Rao, U., Hammen, C., Ortiz, L. R., Chen, L., & Poland, R. E. (2008). Effects of early and recent adverse experiences on adrenal response to psychosocial stress in depressed adolescents. Biological Psychiatry, 64, 521–526.
Snijders, T., & Bosker, R. (1999). Multilevel modeling: An introduction to basic and advanced multilevel modeling. London: Sage.
Stewart, J. G., Mazurka, R., Bond, L., Wynne-Edwards, K. E., & Harkness, K. L. (2013). Rumination and impaired cortisol recovery following a social stressor in adolescent depression. Journal of Abnormal Child Psychology, 41, 1015–1026.
Tanner, J. M. (1962). Growth and adolescence (2nd ed.). Oxford: Blackwell Scientific.
Taylor, S. J., Whincup, P. H., Hindmarsh, P. C., Lampe, F., Odoki, K., & Cook, D. G. (2001). Performance of a new pubertal self-assessment questionnaire: a preliminary study. Paediatric and Perinatal Epidemiology, 15, 88–94.
van Eck, M., Berkhof, H., Nicolson, N., & Sulon, J. (1996). The effects of perceived stress, traits, mood states, and stressful daily events on salivary cortisol. Psychosomatic Medicine, 58, 447–458.
Watson, D., & Clark, L. A. (1991). The mood and anxiety symptom questionnaire. Unpublished Manuscript, Southern Methodist University, Dallas, TX.
Weissman, M. M., Wolk, S., Goldstein, R. B., Moreau, D., Adams, P., Greenwald, S., & Wickramaratne, P. (1999). Depressed adolescents grown up. JAMA, 281, 1707–1713.
Williamson, D. E., Birmaher, B., Frank, E., Anderson, B. P., Matty, M. K., & Kupfer, D. J. (1998). Nature of life events and difficulties in depressed adolescents. Journal of the American Academy of Child and Adolescent Psychiatry, 37, 1049–1057.
Zisook, S., Lesser, I., Stewart, J. W., Wisniewski, S. R., Balasubramani, G. K., Fava, M., & Rush, A. J. (2007). Effect of age at onset on the course of major depressive disorder. American Journal of Psychiatry, 164, 1539–1546.
Acknowledgments
This work was supported by grants from the Ontario Mental Health Foundation (K.H., K.W-E.); as well as a generous donation from Bombardier (K.H.). We gratefully acknowledge Lea Bond for performing the cortisol assays. We would also like to acknowledge Eric Bulmash, Alexandra Sutherland, Nazanin Alavi, Cherie La Rocque, Dustin Washburn, and Jeremy Stewart who performed the diagnostic and life event interviews, and Lindsey Lytle, Shannon Coyle, Jordan Theriault, Monica Haberl, and Chloe Hudson for their help with data coding and management.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This study was approved by the Health Sciences Research Ethics Board at Queen’s University and all study procedures contributing to this work comply with Canadian ethical standards for research involving human participants. Written informed consent was obtained from all participants, and from a parent or guardian for participants under 18 years of age.
Rights and permissions
About this article

Cite this article
Mazurka, R., Wynne-Edwards, K.E. & Harkness, K.L. Stressful Life Events Prior to Depression Onset and the Cortisol Response to Stress in Youth with First Onset Versus Recurrent Depression. J Abnorm Child Psychol 44, 1173–1184 (2016). https://doi.org/10.1007/s10802-015-0103-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10802-015-0103-y