
Abstract
Flexible and free-standing thin films were fabricated and employed directly as working electrodes for the electrochemical reduction of CO2 in 0.5 N KHCO3 at 25 °C, in which, various sizes of Cu2O nanocubes (~ 27 ± 2, 37 ± 3, 62 ± 4 and 207 ± 3 nm) with different extent of surface oxidation (13, 20, 66, and 64% of Cu(II)) were reinforced on to polyvinyl alcohol/reduced graphene oxide matrix (PVA/rGO/(Cu2O/CuO_X, where, X = without halide, Cl, Br and I). The size of Cu2O nanocubes and their surface oxidation were systematically altered by the addition of 1 mL of 10 mM sodium halides (NaCl, NaBr, and NaI) during the synthesis. Energy-dispersive X-ray spectroscopy mapping displayed the specific adsorption of Cl− ions over the Cu2O surface, whereas Br− and I− ions did not show such behaviour. PVA/rGO/(Cu2O/CuO_Cl) thin film exhibited a low overpotential of 20 mV for CO2 reduction reaction and, ~ 60 and ~ 7 times higher current density at − 0.80 V vs. RHE compared to that of the PVA/rGO/(Cu2O/CuO), PVA/rGO/(Cu2O/CuO_Br) ≈ PVA/rGO/(Cu2O/CuO_I), respectively. Gas chromatography and 1H-NMR analyses confirmed methanol as the single liquid product, with a faradaic efficiency of 63% at − 0.75 V vs. RHE on PVA/rGO/(Cu2O/CuO_Cl) thin film.
Graphic abstrat

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Proliferation of carbon dioxide (CO2) is identified as a major reason accountable for the disruption of our eco-system [1] Therefore, it is necessary to control CO2 emissions into the atmosphere and reduce it through environment-friendly approaches. The conversion of CO2 directly into industrial commodities offers double advantage which is carbon–neutral energy-dense fuel production and mitigation of CO2 levels into the atmosphere [2]. Methanol is one such important industrial commodity [3, 4]. Even though there are several catalysts proposed to convert CO2 to methanol, separation of methanol from several products generated is usually a time consuming, inefficient and non-scalable process [5].
For the large scale production of methanol from CO2, an inexpensive, earth-abundant catalyst with facile and sustainable synthesis procedure, selectivity, and stability are extremely desirable. At commercial levels, methanol is being produced via the hydrogenation process (using syngas, i.e. CO and H2) [6, 7]. However, this process requires a large amount of hydrogen, and high temperatures and pressures. CO2 reduction reaction (CO2RR) using electrocatalysts is an attractive green route to generate beneficial fuels such as hydrogen, methane, and methanol at room temperature and pressure [8]. Several metallic electrodes have been used as the electrocatalysts for CO2RR [9,10,11]. Among all, Cu is an earth-abundant and relatively inexpensive element whose surface exhibits low affinity towards CO impurities/intermediates in the course of CO2RR [12]. Primarily, pure Cu particles have been identified as prominent electrocatalysts for the formation of methane, ethylene, and ethanol [13]. Cuprous oxide (Cu2O) is a p-type semiconductor material that has captivated several interests due to its potential applications in catalysis, gas sensing, and solar energy conversion [14]. Cu(I) species in Cu2O could directly reduce the CO2 to methanol due to the facile adsorption of CO2 [15]. To tune the selectivity and catalytic properties of Cu2O various shapes such as nanocubes, nanospheres, nanocages, and hollow structures with different sizes have been synthesized and employed for numerous applications in electrocatalysis [16,17,18].
Composite of Cu2O with conductive materials (such as carbon nanotubes, carbon cloth, rGO, graphene, and polypyrrole) is a promising strategy to improve its electrical conductivity, electrocatalytic activity, and selectivity. Multiwalled carbon nanotubes (MWCNTs) impregnated Cu2O was evaluated for CO2 reduction to methanol, in which MWCNTs stabilizes Cu(I) at the defects of MWCNTs and prevents the formation of Cu(II) [19]. Lower current densities were observed at higher loadings (40–50 wt. %) of Cu2O on MWCNTs due to the agglomeration of Cu2O, which eventually reduces the effective surface area and active sites. Tang and co-workers reported enhanced stability of Cu2O/rGO composite due to the efficient charge transfer from Cu2O to rGO. They also observed an improved photoelectrocatalytic reduction of CO2 to CO on Cu2O/rGO composite [20]. Cu2O supported gas diffusion electrodes have been investigated for the electrochemical reduction of CO2 in 0.5 M KHCO3 [21]. These electrodes have produced mainly methanol (faradaic efficiency, FE, 42.3%), small quantities of ethanol (FE = 10.1%) and n-propanol (FE = 2.4%), whereas, graphene-supported Cu2O electrode has produced exclusively ethanol with FE of 9.93%. Wu and co-workers studied the electrochemical reduction of CO2 on Cu2O cubes (640 nm) deposited carbon clothes (E-TEK) in 0.5 M NaOH electrolyte [22]. Gas chromatography confirmed methanol as the predominant product on Cu2O cubes deposited carbon cloth. Recently, Chang and co-workers prepared the octahedral shaped Cu2O with low index facets and icosahedra shaped Cu2O particles with high index facets followed by functionalization with polypyrrole on a flexible linen paper (Cu2O(OL-MH)/ppy coated LT paper) for the selective formation of methanol (FE = 93%) from CO2RR [23].
Salazar-Villapando and co-workers observed the enhanced current densities during the CO2RR by adding KCl, KBr and KI salts into the aqueous electrolytes [24]. Even though they haven’t analyzed the products of CO2RR, the enhanced current densities are ascribed due to facile electron transfer from Cu-halides to the vacant orbital of CO2. Strasser and Co-workers demonstrated the effect of adding the halides (Cl−, Br− and I−) to the electrolyte on the electrocatalytic activity and selectivity of Cu electrode in CO2RR [25]. The presence of Cl− and Br− aided the increased selectivity of CO formation, while the I− increased the methane production. The effect was ascertained to the high transfer of negative charge to the Cu surface, which eventually favors the protonation of CO to form methane.
Here, we propose the fabrication of scalable free-standing thin films as working electrodes for the electrochemical reduction of CO2 to methanol, in which, surface oxidized Cu2O nanocubes (Cu2O/CuO, Cu2O/CuO_X, where X = Cl, Br, I) were supported on polyvinyl alcohol/rGO (rGO/PVA) matrix (PVA/rGO/(Cu2O/CuO_X)). The fabrication of films on the inexpensive microscopic glass slide was conducted at ambient conditions. Various sizes of copper oxide nanocubes (~ 27 ± 2, 37 ± 3, 62 ± 4 and 207 ± 3 nm) and different extent of surface oxidation (CuO formation) were formed due to the addition of sodium halides (NaCl, NaBr, and NaI) during the synthesis. PVA interlocks rGO via hydrogen bonding and directs the formation of flexible thin films, while Cu2O/CuO nanocubes are anchored into the matrix of PVA/rGO [26, 27]. The free-standing film, PVA/rGO/(Cu2O/CuO_Cl) prepared using Cu2O/CuO_Cl (~ 27 nm) displays ~ 60 and ~ 7 times higher current density compared to that of the PVA/rGO/(Cu2O/CuO), PVA/rGO/(Cu2O/CuO_Br) ≈ PVA/rGO/(Cu2O/CuO_I) films at − 0.80 V vs. RHE, respectively. To achieve the optimum CO2RR activity, the thickness of the films and loading of Cu2O/CuO_Cl were studied by varying the volumes of PVA/rGO/(Cu2O/CuO_Cl) and Cu2O/CuO_Cl solutions respectively.
2 Experimental section
2.1 Materials
Graphene oxide (GO) dispersed in water (5 g L−1) was obtained from UniRegion Bio-Tech (Hsinchu, Taiwan). Hydrazine monohydrate (98%) and potassium hydrogen carbonate (KHCO3) were purchased from Alfa Aesar (Ward Hill, MA, USA). 15 mm length and 6 mm diameter Graphite rod was purchased from Alfa Aesar, USA. Fructose (99%), PVA (average molecular weight: 10,000) and sodium chloride were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ascorbic acid (≥ 99%), Cu(NO3)2‧3H2O (99%), sodium bromide (99.5%), and sodium iodide (99%) were purchased from Acros Organics (Geel, Belgium). Ultrapure water (18.2 MΩ cm) was obtained using a Milli-Q ultrapure system from Merck Millipore (Billerica, MA, USA). Dimethyl sulfoxide unhydrous (DMSO) (> 99.9%) and D2O (99.9 at.% D) solvents were procured from Sigma-Aldrich, USA.
2.2 Methods
X-ray diffraction (XRD) patterns of thin films were recorded using Bruker-8 fitted with Cu Kα radiation (λ = 1.54056 Å). Zeta-potential of PVA/rGO, Cu2O/CuO and Cu2O/CuO_X nanocubes were measured using Zetasizer Nano ZS from Malvern Panalytical, United Kingdom. Raman spectrum of GO, rGO, and PVA/rGO films were obtained using Dongwoo Optron, KyungGiDo, Korea (532 nm laser source) at an accumulation time of 5 min. Keithly 2100/220 digital multimeter, USA was used to measure the conductivity in two probe method. Hitachi S-2400 (Hitachi High-Technologies, Tokyo, Japan) scanning electron microscope (SEM) was used to measure the thickness of PVA/rGO/(Cu2O/CuO_Cl) films. High-resolution transmission electron microscope (HR-TEM), JSM-1200EX II was employed to measure the size of Cu2O/CuO, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I nanocubes as well as their elemental mapping. A VG ECSA210 electron microscope from VG scientific (West Sussex, UK) was employed for X-ray photoelectron spectroscopy (XPS) measurements of various copper-based nanocubes. The electrochemical analysis was performed using a CHI 760D from CH Instruments (Austin, TX, USA). An HP 6890 series Gas Chromatography system fitted with an HP 5973 Mass Selective Detector from Agilent Technologies, Inc. (Wilmington, Delaware, USA) was used to analyze the methanol formed during CO2RR. Prior to acquiring the chromatograms, the column and inlet temperatures were set at 60 and 170 °C, respectively. The gas phase (H2 gas) product was detected by GC-Agilent 7890B equipped with a Restek ShinCarbon ST100/200 mesh (2.0 m in length, 1/16 inch outer diameter and 1.0 mm inner diameter) and operated at oven temperature of 40 °C with N2 as carrier gas. The H2 gas was detected by thermal conductivity detector (TCD). The CO2RR experiment was conducted for an hour in an airtight 2 mL reaction vessel to quantify the gas and liquid products at − 0.75, − 0.80 and − 0.85 V vs. RHE. The required amount of 0.5 M KHCO3 electrolyte (500 μL) was collected from the 2 mL headspace reaction vessel by gas-tight syringe and injected to GC instrument. AVIII600, Bruker Avance™ Nuclear Magnetic Resonance (NMR) spectrometer was used to detect and quantify the methanol amounts. 10 mM of DMSO was added in to the D2O solvent and used as internal standard solution for the 1H-NMR.
2.3 Cu2O/CuO and Cu2O/CuO_X (where X = Cl, Br, I) nanocubes synthesis
The synthesis of Cu2O nanocubes was carried out based on a previously reported work with slight modifications [28]. In addition, three different Cu2O nanocubes were synthesized using NaCl, NaBr and NaI solutions. Briefly, 2.5 mL of 0.01 M Cu(NO3)2 3H2O, and 1.2 g of fructose were added into a 200 mL of double neck round bottom flask, followed by the addition of 2.5 mL of 1 M NaOH and 5 mL of 0.0788 M L-ascorbic acid. The resultant solution was diluted to 100 mL with ultrapure water. Thus prepared solution (pH 12.2) was mildly stirred for 1 h at room temperature. The resulting solution was centrifuged at a relative centrifugal force (RCF) of 20,000×g for 20 min and re-suspended in ultrapure water. The same centrifugation procedure was repeated about three times to remove the unreacted species. The Cu2O nanoparticle solutions were stored at − 4 °C to avoid over-oxidation of the surface. However, partial oxidation of surface is inevitable due to the atmospheric oxygen. A similar procedure was followed by the addition of 1 mL of 10 mM NaCl, NaBr, or NaI aqueous solutions after the L-ascorbic acid. Thus synthesized copper oxide nanocubes are referred to as Cu2O/CuO, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I respectively. Each 1 mL of Cu2O/CuO and Cu2O/CuO_X solutions were dried at 60 °C to determine the weight of Cu2O/CuO. The weight of nanocubes in 1 mL of Cu2O/CuO, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I was found to be 45 ± 3, 42, ± 4, 30 ± 6 and 35 ± 5 µg, respectively.
2.4 Preparation of PVA/rGO composite
10 mL of 5 wt.% PVA aqueous solution was added to 10 mL (50 mg) of GO aqueous solution. Thus resulted PVA-GO mixture was sonicated for 30 min to achieve a homogeneous dispersion. 0.5 mL of hydrazine was added to the PVA-GO solution and heated at 95 °C for 3 h to form the PVA/rGO composite. The resulting solution was cooled and stored at ambient conditions for further use.
2.5 Synthesis of free-standing PVA/rGO/(Cu2O/CuO or Cu2O/CuO_X) thin films
PVA/rGO/(Cu2O/CuO or Cu2O/CuO_X) ink like dispersion was prepared by adding 1.31 µg of Cu2O/CuO or Cu2O/CuO_X solution to PVA/rGO solution containing 1 mg of rGO, followed by 1 h of sonication. Thus resulted ink was transferred to (5 × 2) cm2 area of a microscopic glass mold and dried overnight for solvent evaporation at room temperature. The drying process led to the formation of a thin film, which was carefully peeled off from the glass mold and stored at − 4 °C to avoid the further oxidation of the nanocubes. Cu2O/CuO_Cl loadings (\(\upmu {\text{g}_{{\text{Cu}_{2}}{\text{O}}}/{\text{CuO}}}\_{\text{Cl}}/{\text{mg}}_{{{\text{rGO}}}}\)) and free-standing film thickness (mgrGO/cm2) were optimized to achieve high CO2RR activity. Without incorporating any physical or mechanical support and time-consuming post modifications, all electrochemical measurements were conducted directly on thin films.
2.6 Electrochemical measurements
Electrochemical CO2RR was performed using PVA/rGO/(Cu2O/CuO or Cu2O/CuO_X) thin films as a working electrode, Ag/AgCl (sat. KCl) electrode as a reference electrode and graphite rod as a counter electrode. The working area of the film was maintained to 1 cm2. All potentials measured against the Ag/AgCl reference electrode were then converted to a reversible hydrogen electrode (RHE). The electrolyte was saturated with CO2 by purging the CO2 gas (30 mL/min) into the electrolyte for about 30 min. The pH of the CO2-saturated 0.5 M KHCO3 aqueous solution was determined to be 7.6. During the electrochemical measurements, CO2 gas was continuously purged above the electrolyte. Cyclic voltammetry (CV) was performed to monitor the electrolysis of CO2 using a CHI 760D electrochemical workstation, which was operated in the potential range between 0.68 and − 1.18 V vs. RHE at a scan rate of 20 mV s−1. The amount of methanol formed was determined using a calibration plot obtained from the peak area against the concentration of standard methanol in 0.5 M KHCO3 solutions.
3 Results and discussion
3.1 Formation and characterization of free-standing PVA/rGO/(Cu2O/CuO or Cu2O/CuO_X) thin films
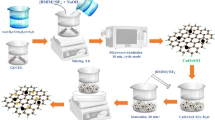
Successful preparation of Cu2O nanocubes was achieved only in alkaline (pH 12.2) conditions by using Cu(NO3)2 3H2O, fructose and ascorbic acid. The Cu2+ ions were stabilized by fructose via Cu(OH)-fructose complexation followed by the reduction to Cu2O. However, considering the partial surface oxidation of Cu2O, the nanocubes were designated as Cu2O/CuO. The growth rate, size, and morphology of Cu2O can be tuned by the presence of halides in the solution, where different halides possess different adsorption strengths on the crystal facets of Cu2O [29]. The addition of 1 mL of 10 mM sodium halides (NaCl, NaBr, or NaI) during the synthesis of nanocubes alters the ionic strength of the solution, which affects the growth of Cu adatoms and hence the size of nanocubes [30, 31]. Scheme 1a illustrates the step-wise processes involved in the synthesis of Cu2O/CuO nanocubes. Figure 1a–d displays HR-TEM images of Cu2O/CuO, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I nanocubes, respectively. The average size of Cu2O/CuO nanocubes was calculated to be 37 ± 3 nm, while the presence of Cl− ions led to the formation of 10 nm smaller Cu2O/CuO_Cl nanocubes. While conducting the same synthesis procedure in presence of Br− or I−, larger particles were formed with average diameters of 62 ± 4 and 207 ± 3 nm, respectively. Figure S1(A) shows the energy dispersive X-ray spectroscopy (EDS) mapping of Cu, Cl−, Br− and I− elements. Figure S1(B) displays the EDS spectrum of the Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I nanocubes. The EDS mapping unambiguously displays the spatial distribution of Cl− ions with at.% of 0.39 on the surface of nanocubes, confirming the specificity of Cl− ion adsorption over the surface of Cu2O. Hence, the growth of Cu2O cubes was ceased by the preferable adsorption of Cl− ions over the surface of Cu2O, which resulted in smaller nanocubes. The adsorbed Cl− ions thermodynamically stabilizes the surface structures and increases the surface area of Cu2O/CuO_Cl [29, 32]. Random and lower intensities of purple dots were observed in Figure S1(A)b″ and c″, which indicates the presence Br− and I− ions without any specific adsorption on the surface of nanocubes. In addition, the at.% of Cl (0.39%) is higher than the Br− (0.20%) and I− (0.31%) elements. Ignaczak et al. has shown that the decrease in adsorption energy while going from F− to I− on the Cu, Ag and Au metals via density functional theory calculations [33]. XPS is a sensitive technique to gain insight into the surface composition and electronic state of the species. The surface of Cu2O could undergo oxidation in ambient conditions and leave the surface of nanocubes with mixed oxidation states of Cu (such as Cu+ and Cu2+) [34]. Quantification of the relative amount of Cu(II) species on the Cu2O surface could give an insight into the catalytic activity of Cu2O nanocubes [35]. The ratios of Cu (I): Cu(II) was calculated from the Cu2p3/2 spectra using the methodology developed by Gerson and Jaseniak [36]. Figure 2 shows the Cu 2p core level profile XPS spectra of as-synthesized (a) Cu2O/CuO, (b) Cu2O/CuO_Cl, (c) Cu2O/CuO_Br and (d) Cu2O/CuO_I nanocubes. The deconvoluted XPS spectra of (a), (b), (c) and (d) displays the Cu2p3/2 level at 934.3, 934.2, 933.1 and 933.0 eV; Cu2p1/2 level at 954.3, 954.3, 954.0 and 952.8 eV respectively, which can be assigned to Cu+ present in Cu2O. Particularly, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I nanocubes show peaks at 936.9, 935.0 and 934.9 eV respectively, these peaks can be assigned to Cu2+, which indicates the formation of CuO surface layer. The main peak present (935.0 and 934.9 eV) in Cu2O/CuO_Br and Cu2O/CuO_I nanocubes indicates the significant oxide layer over the Cu2O surface. Hence, the availability of Cu2O active sites to the CO2RR has diminished significantly. Cu2O/CuO_Cl nanocubes consist of mainly Cu2O species (80%) with a minor surface CuO phase (20%). Peaks at 944.1, 944.5, 941.0, 942.0; 949.9 eV were identified as the satellite peaks of CuO Cu2p3/2 and 963.8, 963.9, 963.5 and 963.5 eV were identified as the satellite peaks of CuO Cu2p1/2 level. The ratios of Cu2O: CuO for (a) Cu2O/CuO, (b) Cu2O/CuO_Cl, (c) Cu2O/CuO_Br and (d) Cu2O/CuO_I was calculated to be 87:13, 80:20, 34:66 and 35:65.

Schematic representation of (A) synthesis of (a) Cu2O/CuO_Cl, (b) Cu2O/CuO_Br, (c) Cu2O/CuO_I, nanocubes, (B) preparation of PVA/rGO followed by PVA/rGO/(Cu2O/CuO) composite

HR-TEM images, a Cu2O/CuO, b Cu2O/CuO_Cl, c Cu2O/CuO_Br and d Cu2O/CuO_I nanocubes, inset graphs represents the size distribution of corresponding nanocubes, respectively.

Deconvoluted Cu 2p core level region X-ray photoelectron spectra of a Cu2O/Cuo, b Cu2O/CuO_Cl, c Cu2O/CuO_Br and d Cu2O/CuO_I nanocubes (‘s’ stands for satellite peak)
PVA is a polymer containing hydroxyl functional groups with excellent chemical stability and film-forming ability [37]. Scheme 1b shows the synthesis of PVA/rGO and PVA/rGO/(Cu2O/CuO) composites. An equal volume ratio of 5 wt.% PVA and GO aqueous solutions were thoroughly mixed to obtain a PVA-GO solution. The hydroxyl groups in PVA interact with each other via intra-molecular hydrogen bonding. GO could interact with the hydroxyl groups of PVA via intermolecular hydrogen bonding, which is then reduced by hydrazine at 90 °C for 3 h to form a macroscopically homogeneous PVA/rGO solution [26]. The brown color of the PVA-GO solution, slowly converted to black, indicating the formation of rGO. Raman spectroscopy was employed to confirm the formation of rGO from GO. Figure 3a shows the comparison of Raman spectra of GO, rGO, and PVA/rGO composites. D- and G-band of all the three samples were observed at 1346 and 1582 cm−1, respectively. The D- and G-bands originated from the first-order scattering of E2g vibrational modes in the graphitic forms (sp2-carbon) and defects (sp3-carbon), respectively. In the Raman spectrum of rGO and PVA/rGO, D-band intensity decreased while G-band becomes prominent. D-band to G-band ratio (D/G ratio) for GO, rGO and PVA/rGO is found to be 1.451, 0.851 and 0.671, respectively. The decrease in the D/G ratio of PVA/rGO composite confirms the formation of rGO and sp2 networks on the basal planes through the hydrazine reduction [38].

(A) Raman spectra of GO, rGO and PVA/rGO films, and (B) XRD pattern of (a) PVA/rGO/(Cu2O/CuO), (b) PVA/rGO/(Cu2O/CuO_Cl), (c) PVA/rGO/(Cu2O/CuO_Br) and (d) PVA/rGO/(Cu2O/CuO_I) films, respectively.
The electrostatic interactions between the Cu2O and PVA/rGO are one of the possible forces governing the affinity of nanocubes into the network rGO/PVA matrix. Hence, ζ-potential measurements were conducted to assess the electrostatic interactions between PVA/rGO and Cu2O/CuO or Cu2O/CuO_X nanocubes. Table 1 shows the ζ-potential values for the aqueous solutions of PVA/rGO, Cu2O/CuO, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I nanocubes along with their corresponding standard deviation. The ζ-potential of PVA/rGO was found to be − 6.34 mV with a standard deviation of ± 0.14 mV. The negative ζ-potential value indicates the presence of anionic sites over the PVA/rGO matrix for the adsorption of cationic species. However, ζ-potential values for Cu2O/CuO, Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I nanocubes were also observed to be negative which were − 19.16, − 13.50, − 38.43 and − 36.26 mV, respectively. The nanocubes which posses the high negative ζ-potential will be repelled stronger from PVA/rGO matrix and hence, weaker attractive forces are expected. The weak electrostatic attractions would operate between PVA/rGO and Cu2O/CuO, Cu2O/CuO_Br, and Cu2O/CuO_I compared to that of the PVA/rGO and Cu2O/CuO_Cl nanocubes. The relatively stronger columbic interaction of Cu2O/CuO_Cl nanocubes with the PVA/rGO matrix could lead to the least charge transfer resistance, hence the higher CO2RR activity. To accomplish the optimum CO2RR activity, the thickness of the films and loading of Cu2O/CuO_Cl were studied by varying the volumes of PVA/rGO/(Cu2O/CuO_Cl) and Cu2O/CuO_Cl solutions, respectively.
The preparation of the thin films on microscopic slides is schematically represented in Scheme S1. PVA/rGO/(Cu2O/CuO) solution was sonicated for 1 h to form an ink-like dispersion. The resulting ink (1 mL) was dropped on to (5 × 2) cm2 area of a microscopic glass slide and dried overnight at room temperature to form PVA/rGO/(Cu2O/CuO) film. Similarly, PVA/rGO/(Cu2O/CuO_Cl), PVA/rGO/(Cu2O/CuO_Br), and PVA/rGO/(Cu2O/CuO_I), thin films were prepared using ~ 1.31 μg of Cu2O/CuO_Cl, Cu2O/CuO_Br, and Cu2O/CuO_I solutions, respectively. In Fig. 3b, XRD patterns of all the films displayed a broad peak at 2θ value of 19.5° which corresponds to PVA [39]. The characteristic peaks of Cu2O/CuO or Cu2O/CuO_X did not appear because of their ultra-low (0.87–3.06 μg/cm2) loadings and their existence within the PVA/rGO sheets. The mechanical properties of PVA fibers facilitate the formation of a stable and flexible film. The free-standing PVA/rGO/(Cu2O/CuO_Cl) thin film is stable after twisting, and tape tests, showing its mechanical strength which is one of the desired properties of free-standing working electrodes (Figure S2). In the case of free-standing electrodes, no direct support of the current collector is used; hence the intrinsic electrical conductivity of thin films itself needs to be high enough to conduct the electrons. Electrical conductivity of PVA/rGO/(Cu2O/CuO), PVA/rGO/(Cu2O/CuO_Cl), PVA/rGO/(Cu2O/CuO_Br), and PVA/rGO/(Cu2O/CuO_I), films were measured to be 0.160, 0.182, 0.185 and 0.380 S/m via two probe-method, respectively. These conductivity values are four orders of magnitude lower than that of the reported rGO (2420 S/m) and 30 times lower than the rGO/PVA [39, 40]. The presence of non-conducting PVA on to the rGO sheets could decrease the conductivity of the free-standing thin films.
3.2 Electrochemical reduction of CO2
Figure 4a shows the CVs of PVA/rGO/(Cu2O/CuO_Cl) film recorded in the N2-saturated and CO2-saturated 0.5 M KHCO3 solution. The enhanced current density exhibited in CO2-saturated 0.5 M KHCO3 electrolyte indicates the electrocatalytic ability of PVA/rGO/(Cu2O/CuO_Cl) thin film for CO2RR. Figure 4b shows CVs of free-standing films of PVA/rGO/(Cu2O/CuO), PVA/rGO/(Cu2O/CuO_Cl), PVA/rGO/(Cu2O/CuO_Br), and PVA/rGO/(Cu2O/CuO_I), prepared from different sized Cu2O/CuO_X nanocubes, recorded at a scan rate of 20 mV s−1 in CO2-saturated 0.5 M KHCO3 solution at 25 °C. The PVA/rGO/(Cu2O/CuO_Cl) thin film displayed a remarkable CO2RR activity, in terms of low onset potential and higher current densities in comparison to that of PVA/rGO/(Cu2O/CuO), PVA/rGO/(Cu2O/CuO_Br), and PVA/rGO/(Cu2O/CuO_I) thin films. PVA/rGO/(Cu2O/CuO_Cl) film showed an onset potential of − 0.40 V vs. RHE for CO2RR, which is much lower than that of PVA/rGO/(Cu2O/CuO), (− 0.43 V vs. RHE), PVA/rGO/(Cu2O/CuO_Br) (− 0.80 V vs. RHE) and PVA/rGO/(Cu2O/CuO_I), (− 0.66 V vs. RHE) films. It exhibited a current density of 0.12 mA cm−2 at − 0.80 V vs. RHE, which is ~ 7 times higher than that of the other films studied for CO2RR (see Figure S3). The superior CO2RR activity of PVA/rGO/(Cu2O/CuO_Cl) thin film is likely due to a large number of active sites and relatively smaller sizes of Cu2O nanocubes. Cl− ions could suppress the unwanted hydrogen product, which increases the CO2RR activity and selectivity of PVA/rGO/(Cu2O/CuO_Cl) thin film [32, 41].

Comparison of cyclic voltammograms of A PVA/rGO/(Cu2O/CuO_Cl) film in N2−, CO2-saturated 0.5 M KHCO3, B (a) PVA/rGO/(Cu2O/CuO), (b) PVA/rGO/(Cu2O/CuO_Cl), (c) PVA/rGO/(Cu2O/CuO_Br) and (d) PVA/rGO/(Cu2O/CuO_I) films in CO2-saturated 0.5 M KHCO3 (pH 7.6) at the scan rate of 20 mV s−1, 25 °C; C comparison of gas chromatograms of standard (std.) solution (250 µM methanol in 0.5 M KHCO3) and 0.5 M KHCO3 electrolyte collected after 1 h of CO2RR on PVA/rGO/(Cu2O/CuO_Cl) film at (a) − 0.85, (b) − 0.80 and (c) − 0.75 V vs. RHE; D methanol formation rates (µmol cm−2 h−1) determined at different potentials for the film (a).
Further, CO2RR activity of PVA/rGO/(Cu2O/CuO_Cl) thin film was optimized with respect to Cu2O/CuO loadings (\(\upmu {\text{g}_{{\text{Cu}_{2}}{\text{O}}}/{\text{CuO}}}\_{\text{Cl}}/{\text{mg}}_{{{\text{rGO}}}}\)). By using aliquots of 100–350 µL) of Cu2O/CuO_Cl solutions, various amounts (0.87–3.06 µg) of Cu2O/CuO_Cl were loaded onto each one mg of PVA/rGO composite. Figure S4(B) shows the CO2RR activity of PVA/rGO/(Cu2O/CuO_Cl) film at various loadings of Cu2O/CuO-Cl nanocubes. Upon increasing the Cu2O/CuO_Cl loadings from 0.87 to 2.19 µg, an increase in current density and a decrease in the onset potential were noted. Further increase in Cu2O loading to 3.12 µg resulted in poor CO2RR activity, revealing that higher Cu2O loadings could lead to aggregation of particles that cause the lower number of active sites for CO2RR. Thus, optimum CO2RR activity was found at a Cu2O/CuO_Cl loading of 1.31 µg. We then optimized the film thickness by changing the rGO loading per unit area to further enhance CO2RR activity. Figure S4(A) shows the CO2RR activity of films prepared at different loadings of rGO. High rGO loadings could decrease the surface area, deform the regular arrangements of PVA chains of PVA/rGO/(Cu2O/CuO_Cl) films which led to the poor CO2RR activity [42]. The thin film prepared using 0.492 mg of rGO showed the optimum CO2RR activity. Figure S5 shows the cross-sectional SEM images of the respective films prepared with different rGO loadings. The thickness of the films prepared using the rGO loadings of 0.246, 0.492, 0.738 and 0.984 mg were 11, 14, 16, and 32 µm, respectively.
3.3 Analysis of products
Chronoamperometry was used to monitor the product of electrolysis of CO2 in the potential range between − 0.75 to − 0.85 V vs. RHE for 1 h. In order to identify and quantify the products during the course of CO2RR, controlled potential electrolysis was carried out at potentials of − 0.75, − 0.80 and − 0.85 V vs. RHE for 1 h in 0.5 M KHCO3 solution. GC–MS was used to analyze the liquid products formed during the CO2RR. Figure 4c shows the GC spectra of standard methanol solution and that of the solutions subjected to electrolysis of CO2 at different potentials using the free-standing PVA/rGO/(Cu2O/CuO_Cl) thin film. A sharp peak corresponding to methanol was observed at a retention time of 2.54 min. We observed only methanol as a product in the liquid phase, which confirms the high selectivity of the PVA/rGO/(Cu2O/CuO_Cl) thin film towards methanol formation in CO2RR. The amount of methanol formed was then determined using a calibration curve of the peak area against the concentration of standard methanol solution in 0.5 M KHCO3 (Figure S6). The amounts of methanol formed at the potentials of − 0.75, − 0.80 and − 0.85 V vs. RHE for 1 h were found to be 27, 35 and 39 µmol, respectively (Fig. 4d). The highest methanol yield of 19.5 µmol cm−2 h−1 was observed at − 0.85 V vs. RHE. Le et al. have reported the methanol yield of 43 µmol cm−2 h−1 at − 1.32 V vs. RHE for electrodeposited Cu2O in 0.5 M KHCO3, however, the onset potential for the CO2RR was observed at high overpotentials (− 0.75 V vs. RHE) [43]. Methanol yield and onset potential of PVA/rGO/(Cu2O/CuO_Cl) film for CO2RR are superior to that of the several catalysts reported in the literature (Table 2) [44,45,46,47,48]. The CO2RR experiment was conducted using chronoamperometric technique (− 0.75, − 0.80 and − 0.85 V vs. RHE) in a sealed 2 mL reaction vessel to quantify the gas and liquid products in CO2 saturated 0.5 M KHCO3. Figure S7 displays the i–t curves for CO2RR on PVA/rGO/(Cu2O/CuO_Cl) in 0.5 M KHCO3 at − 0.75, − 0.80 and − 0.85 V vs. RHE. Figure S8 represents the calibration plot for H2 gas. The GC calibration line for H2 gas was obtained by injecting the known volumes of H2 from 2 to 25 μL corresponding to 0.1 to 1.2 umol of H2 gas, respectively. A 500 μL of gas was collected from the 2 mL headspace reaction vessel after 1 h of CO2RR using gas-tight syringe followed by sample injection to GC instrument. Figure S9 shows the GC plot obtained at (A) − 0.75, (B) − 0.80, and (C) − 0.85 V vs. RHE. The amount of H2 gas was calculated to be 1.60 × 10–6, 0.92 × 10–6 and 2 × 10–6 μmol cm−2 at − 0.75, − 0.80 and − 0.85 V vs. RHE, respectively. The FE for the H2 was calculated to be 20, 14 and 32% at − 0.75, − 0.80 and − 0.85 V vs. RHE, respectively. The amount of methanol was quantified using 1H-NMR at − 0.75, − 0.80 and − 0.85 V vs. RHE. A 35 μL of standard solution and 500 μL of 0.5 M KHCO3 was mixed thoroughly and analyzed with AVIII600 NMR. Figure 5 shows the 1H-NMR of the electrolyte which was collected after 1 h of CO2RR at different potentials. The 1H-NMR peaks around 4.59, 3.17 and 2.5 ppm indicate the H2O, methanol and DMSO, respectively. The Polyvinyl alcohol, which is employed in the thin film electrodes preparation, was dissolved in to the electrolyte. The ‘methene’ (2) and ‘methine’(1) protons were appeared at 3.42 and 0.9 ppm, respectively [49]. The FE for the methanol was found to be 63, 43 and 59% at − 0.75, − 0.80 and − 0.85 V vs. RHE, respectively. Table S1 lists the amount and FE for methanol and H2 gas. The FE % is higher than several reported catalysts for methanol production in CO2RR [21, 46, 48, 50]. Figure 6 displays the FE % for the methanol and H2 gas in CO2RR on PVA/rGO/(Cu2O/CuO_Cl). The durability in methanol production of PVA/rGO/(Cu2O/CuO_Cl) in 0.5 M KHCO3 was evaluated for 48 h. 1 mL of 0.5 M KHCO3 was collected from 25 mL of electrolyte for sampling at respective times followed by introducing 1 mL of CO2 saturated 0.5 M KHCO3. Figure S10 (A) shows the chronoamperometric graph for 48 h and (B) 1H-NMR data shows the methanol as a liquid product. The amount of methanol was found to be 18, 20, 93 and 77 μmol cm−2 for 1, 5, 30 and 48 h duration, on PVA/rGO/(Cu2O/CuO_Cl) at − 0.75 V vs. RHE, respectively (Fig. 7).

1H-NMR of the liquid products in CO2RR on PVA/rGO/(Cu2O/CuO_Cl) (10 mM DMSO added as an internal standard in D2O solvent)

Faradaic efficiency for methanol and H2 gas in CO2RR on PVA/rGO/(Cu2O/CuO_Cl) thin film electrode in 0.5 M KHCO3 at − 0.75, − 0.80 and − 0.85 V vs. RHE about 1 h

The methanol production rates at 1, 5, 30 and 48 h of CO2RR on PVA/rGO/(Cu2O/CuO_Cl) thin film electrode in 0.5 M KHCO3 electrolyte at − 0.75 vs. RHE
Cross-linked PVA with rGO could facilitate the permeability of CO2 molecules through film [51, 52]. PVA molecules show excellent mechanical strength to the film and also acts as supporting units for rGO [53,54,55,56]. Partially oxidized Cu2O nanocubes provide the active sites for CO2 reduction, whereas PVA/rGO matrix enhances the electron transport in the film. Recently, Periasamy et al. have reported that methanol is formed on the Cu2O surface through the electrochemical reduction of CO2,ads to CO, followed by the hydrogenation of CO to CH3O−, which reduced further to form methanol [23]. Similar reaction pathways and mechanisms are likely to occur on the surfaces of Cu2O nanocubes anchored on the surfaces of PVA/rGO leading to the formation of methanol. Although Strasser et al. have doubled the CO production efficiency of Cu electrodes through the direct addition of halides each at a concentration of 0.3 M into the testing electrolyte, their electrochemical behavior under dynamic and stationary potential conditions was different and thus they were unable to generate a valuable hydrocarbon product (methanol) [25]. Unlike this approach, we directly added relatively fewer amounts of halides (10 mM vs. 0.3 M) into the Cu2O preparation solution and tuned the size of Cu2O particles by controlling the specific adsorption capacity of halide ions. As a result, we were able to control the surface structures, the active sites, the reaction intermediates, and the rate of methanol formation. The Cl− ions adsorbed on the surfaces of Cu2O remarkably accelerates the CO2− formation and enhances the CO production. The negative charge induced by the Cl− ions result in a positive effect leading to a facile protonation/hydrogenation of CO to form CH3O− and thus the methanol [25]. The enhanced electron flow from the Cl− orbital to the vacant CO2 orbital through a nucleophilic attack on the carbon atom of CO2, further enhances the current density, methanol selectivity, and methanol formation rate [57].
4 Conclusions
Direct evaporation assisted self-assembly strategy for the fabrication of free-standing thin films was employed using PVA, rGO and partial surface oxidized Cu2O nanocubes (Cu2O/CuO) on an inexpensive microscopic glass slide. Cu2O/CuO nanocubes were synthesized in the presence of sodium halides (1 mL of 10 mM NaCl, NaBr and NaI) at room temperature. Cl− ion adsorption over the surface of Cu2O hindered the adsorption of adatoms to Cu2O crystal facets and formed 27 ± 2 nm size particles, whereas Br− and I− ions did not show notable adsorption on Cu2O surface and yielded the particles with larger sizes, 62 ± 4 and 207 ± 3 nm, respectively. The high-resolution photoelectron spectrum revealed a small CuO surface phase present over the Cu2O nanocubes. The ratio of Cu2O: CuO for (a) Cu2O/CuO, (b) Cu2O/CuO_Cl, (c) Cu2O/CuO_Br and (d) Cu2O/CuO_I was calculated to be 87:13, 80:20, 34:66 and 35:65. PVA/rGO/(Cu2O/CuO_Cl) film exhibited superior CO2 reduction activity in terms of onset potential (− 0.40 V vs. RHE) and current density (0.12 mA cm−2 at − 0.80 V vs. RHE) than that of the PVA/rGO/(Cu2O/CuO), PVA/rGO/(Cu2O/CuO_Br), and PVA/rGO/(Cu2O/CuO_I) thin films. GC–MS analysis revealed the formation of methanol as a single liquid product. Methanol yield of 19.5 µmol cm−2 h−1 at − 0.85 V vs. RHE is observed on the PVA/rGO/(Cu2O/CuO_Cl) thin film. The FE for methanol and H2 gas were found to be 63 and 20% at − 0.75 V vs RHE, respectively, which is the highest FE among the – 0.75, − 0.80 and − 0.85 V vs. RHE. With the advantages of low-cost, flexibility, high mechanical strength, catalytic activity, and stability, the free-standing thin films prepared through direct evaporation assisted self-assembly method holds great potential for the use in electrocatalysis and chemical industries to generate pure methanol.
References
Anderson TR, Hawkins E et al (2016) CO2, the greenhouse effect and global warming: from the pioneering work of Arrhenius and Callendar to today’s Earth System Models. Endeavour 40(3):178–187
Saeidi S, Amin NAS et al (2014) Hydrogenation of CO2 to value-added products—a review and potential future developments. J CO2 Util 5:66–81
Albo J, Alvarez-Guerra M et al (2015) Towards the electrochemical conversion of carbon dioxide into methanol. Green Chem 17(4):2304–2324
Joghee P, Malik JN et al (2015) A review on direct methanol fuel cells—in the perspective of energy and sustainability. MRS Energy Sustain 2:E3
Ganesh I (2014) Conversion of carbon dioxide into methanol—a potential liquid fuel: Fundamental challenges and opportunities (a review). Renew Sustain Energy Rev 31:221–257
Luu MT, Milani D et al (2015) A comparative study of CO2 utilization in methanol synthesis with various syngas production technologies. J CO2 Util 12:62–76
von der Assen N, Voll P et al (2014) Life cycle assessment of CO2 capture and utilization: a tutorial review. Chem Soc Rev 43(23):7982–7994
Gao D, Cai F, Wang G et al (2017) Nanostructured heterogeneous catalysts for electrochemical reduction of CO2. Curr Opin Green Sustain Chem 3:39–44
Lim RJ, Xie M, Sk MA et al (2014) A review on the electrochemical reduction of CO2 in fuel cells, metal electrodes and molecular catalysts. Catal Today 233:169–180
Qiao J, Liu Y, Hong F et al (2014) A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem Soc Rev 43(2):631–675
Kumar B, Brian JP et al (2016) New trends in the development of heterogeneous catalysts for electrochemical CO2 reduction. Catal Today 270:19–30
Hori Y, Wakebe H et al (1994) Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim Acta 39(11):1833–1839
Raciti D, Wang C (2018) Recent advances in CO2 reduction Electrocatalysis on copper. ACS Energy Lett 3(7):1545–1556
Schreier M, Luo J et al (2016) Covalent immobilization of a molecular catalyst on Cu2O photocathodes for CO2 reduction. J Am Chem Soc 138(6):1938–1946
Peterson AA, Abild-Pedersen F et al (2010) How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ Sci 3(9):1311–1315
Nitopi S, Bertheussen E et al (2019) Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem Rev 119(12):7610–7672
Raciti D, Wang Y et al (2018) Three-dimensional hierarchical copper-based nanostructures as advanced electrocatalysts for CO2 reduction. ACS Appl Energy Mater 1(6):2392–2398
Gu Z, Shen H et al (2018) Nanostructured copper-based electrocatalysts for CO2 reduction. Small Methods 2(11):1800121
Irfan Malik M, Malaibari ZO et al (2016) Electrochemical reduction of CO2 to methanol over MWCNTs impregnated with Cu2O. Chem Eng Sci 152:468–477
An X, Li K et al (2014) Cu2O/reduced graphene oxide composites for the photocatalytic conversion of CO2. Chemsuschem 7(4):1086–1093
Albo J, Irabien A (2016) Cu2O-loaded gas diffusion electrodes for the continuous electrochemical reduction of CO2 to methanol. J Catal 343:232–239
Chang T-Y, Liang R-M et al (2009) Electrochemical reduction of CO2 by Cu2O-catalyzed carbon clothes. Mater Lett 63(12):1001–1003
Periasamy AP, Ravindranath R et al (2018) Facet- and structure-dependent catalytic activity of cuprous oxide/polypyrrole particles towards the efficient reduction of carbon dioxide to methanol. Nanoscale 10(25):11869–11880
Salazar-Villalpando MD (2011) Effect of electrolyte on the electrochemical reduction of CO2. ECS Trans 33(27):77–88
Varela AS, Ju W et al (2016) Tuning the catalytic activity and selectivity of Cu for CO2 electroreduction in the presence of halides. ACS Catal 6(4):2136–2144
Feng H, Li Y et al (2012) Strong reduced graphene oxide–polymer composites: hydrogels and wires. RSC Adv 2(17):6988–6993
Sahoo NG, Pan Y et al (2012) Graphene-based materials for energy conversion. Adv Mater 24(30):4203–4210
Yang Z, Chiang CK et al (2007) Synthesis of fluorescent and photovoltaic Cu2O nanocubes. Nanotechnology 19(2):025604
Tang Z, Kwon H et al (2017) Role of halide ions for controlling morphology of copper nanocrystals in aqueous solution. ChemistrySelect 2(17):4655–4661
Meena SK, Celiksoy S et al (2016) The role of halide ions in the anisotropic growth of gold nanoparticles: a microscopic, atomistic perspective. Phys Chem Chem Phys 18(19):13246–13254
Rai A, Singh A et al (2006) Role of halide ions and temperature on the morphology of biologically synthesized gold nanotriangles. Langmuir 22(2):736–741
Hsieh Y-C, Senanayake SD et al (2015) Effect of chloride anions on the synthesis and enhanced catalytic activity of silver nanocoral electrodes for CO2 electroreduction. ACS Catal 5(9):5349–5356
Ignaczak A, Gomes JANF (1997) Quantum calculations on the adsorption of halide ions on the noble metals. J Electroanal Chem 420(1):71–78
Frese KW (1991) Electrochemical reduction of CO2 at intentionally oxidized copper electrodes. J Electrochem Soc 138(11):3338–3344
Wang M, Ren X et al (2020) Selective electroreduction of CO2 to CO over co-electrodeposited dendritic core-shell indium-doped Cu@Cu2O catalyst. J CO Util 37:204–212
Gerson AR, Jasieniak M (2008) In: Duo WD, Yao SC, Liang WF, Cheng ZL, Long H (eds) Proceedings of the XXIV international minerals processing congress. Science Press, Beijing, pp 1054–1063
Tripathi S, Mehrotra GK et al (2009) Physicochemical and bioactivity of cross-linked chitosan–PVA film for food packaging applications. Int J Biol Macromol 45(4):372–376
Park S, Hu Y et al (2012) Chemical structures of hydrazine-treated graphene oxide and generation of aromatic nitrogen doping. Nat Commun 3(1):638
Yang J-H, Lee Y-D (2012) Highly electrically conductive rGO/PVA composites with a network dispersive nanostructure. J Mater Chem 22(17):8512–8517
Stankovich S, Dikin DA et al (2007) Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 45(7):1558–1565
Quan F, Zhong D et al (2015) A highly efficient zinc catalyst for selective electroreduction of carbon dioxide in aqueous NaCl solution. J Mater Chem A 3(32):16409–16413
Zhao X, Zhang Q et al (2010) Enhanced mechanical properties of graphene-based poly(vinyl alcohol) composites. Macromolecules 43(5):2357–2363
Le M, Ren M et al (2011) Electrochemical reduction of CO2 to CH3OH at copper oxide surfaces. J Electrochem Soc 158(5):E45–E49
Schizodimou A, Kyriacou G (2012) Acceleration of the reduction of carbon dioxide in the presence of multivalent cations. Electrochim Acta 78:171–176
Jia F, Yu X et al (2014) Enhanced selectivity for the electrochemical reduction of CO2 to alcohols in aqueous solution with nanostructured Cu–Au alloy as catalyst. J Power Sources 252:85–89
Albo J, Vallejo D et al (2017) Copper-based metal-organic porous materials for CO2 electrocatalytic reduction to alcohols. Chemsuschem 10(6):1100–1109
Albo J, Beobide G et al (2017) Methanol electrosynthesis from CO2 at Cu2O/ZnO prompted by pyridine-based aqueous solutions. J CO2 Util 18:164–172
Albo J, Sáez A et al (2015) Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl Catal 176–177:709–717
Li B, Gao Y, Guo L, Fan Y, Kawazoe N, Fan H, Zhang X, Chen G (2018) Synthesis of photo-reactive poly (vinyl alcohol) and construction of scaffold-free cartilage like pellets in vitro. Regen Biomater 5(3):159–166
Yuan J, Wang X, Gu C, Sun J, Ding W, Wei J, Zuo X, Hao C (2017) Photoelectrocatalytic reduction of carbon dioxide to methanol at cuprous oxide foam cathode. RSC Adv 7(40):24933–24939
Liu Y, Su Y et al (2018) Asymmetric aerogel membranes with ultrafast water permeation for the separation of oil-in-water emulsion. ACS Appl Mater Interfaces 10(31):26546–26554
Mondal A, Mandal B (2014) Novel CO2-selective cross-linked poly(vinyl alcohol)/polyvinylpyrrolidone blend membrane containing amine carrier for CO2–N2 separation: synthesis, characterization, and gas permeation study. Ind Eng Chem Res 53(51):19736–19746
Wang T, Li Y et al (2015) Preparation of flexible reduced graphene oxide/poly(vinyl alcohol) film with superior microwave absorption properties. RSC Adv 5(108):88958–88964
Zhang X, Zhou Y et al (2017) In-situ reduced graphene oxide-polyvinyl alcohol composite coatings as protective layers on magnesium substrates. Prog Nat Sci Mater Int 27(3):326–328
Kim S, Shimazu J et al (2017) Thermal conductivity of graphene oxide-enhanced polyvinyl alcohol composites depending on molecular interaction. Polymer 129:201–206
Xu Y, Hong W et al (2009) Strong and ductile poly(vinyl alcohol)/graphene oxide composite films with a layered structure. Carbon 47(15):3538–3543
Ogura K, Salazar-Villalpando MD (2011) CO2 electrochemical reduction via adsorbed halide anions. JOM 63(1):35–38
Acknowledgements
RS and AS thanks to the MOST (Ministry Of Science and Technology), Taiwan for financial support under project number MOST-108-2112-M-001-049-MY2. We also thank Ms. S.-J. Ji and C.-Y. Chien of the Precious Instrument Center (National Taiwan University, Taiwan) for their assistance in SEM. The assistance of Ms. S.-J. Ji and C.-Y. Chien from the Instrument Center at NTU for TEM measurement is appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sheelam, A., Muneeb, A., Talukdar, B. et al. Flexible and free-standing polyvinyl alcohol-reduced graphene oxide-Cu2O/CuO thin films for electrochemical reduction of carbon dioxide. J Appl Electrochem 50, 979–991 (2020). https://doi.org/10.1007/s10800-020-01450-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-020-01450-z