
Abstract
Antimony selenide and its carbon composite were synthesized through a mechanochemical process and investigated as anode materials for sodium-ion secondary batteries. X-ray diffraction (XRD) with rietveld refinement and transmission electron microscopy (TEM) analyses confirm that Sb2Se3 were composed of agglomerated highly crystalline nanocrystallites and the Sb2Se3/C composite consisted of nanocrystalline Sb2Se3 dispersed homogeneously throughout an amorphized carbon matrix. The initial Coulombic efficiency, rate capability, and cycle performance of the Sb2Se3/C composite were superior to those of Sb, or Sb2Se3. The Sb2Se3/C composite, in particular, showed excellent cycle stability, with 98.2% of initial capacity at 200 mA g−1 after 200 cycles. Based on the reaction potentials, ex situ XRD patterns and ex situ HR-TEM analysis of the Sb2Se3/C composite electrode revealed the structural changes which occurred reversibly within the Sb2Se3/C composite by conversion and recombination reaction during sodiation and desodiation process. Furthermore, XPS analysis study was carried out for identifying the surface films formed on both the electrodes and their effects on the performances.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Lithium-ion batteries (LIBs) are commonly used as the main power sources in portable electronic devices, such as cellular phones, laptops, and tablet PCs, because LIBs perform well in terms of volumetric/gravimetric energy density, cycle life, and reliability compared to other types of battery systems [1, 2]. In recent years, as the demands on power sources in large-scale energy storage systems (ESSs) and electric vehicles (EVs) have increased, the applications of LIBs have expanded from mobile electronic devices to large-scale power devices [3, 4]. Although LIBs are very attractive as power sources in ESSs and EVs, lithium reserves are limited, concentrated in remote or politically troubled areas such as South Africa, and unable to respond to an increase in the global scale demand [5]. In addition, the costs associated with extracting the carbonate precursor needed to produce lithium are expected to increase as the demand for LIBs increases. Current efforts seek to develop other types of battery systems not encumbered by costs or resource limitations and with energy density or cycle life performances that are comparable to those of LIB.
Na-ion batteries (NIBs) have attracted attention as an alternative to LIBs because sodium is abundant, and its extraction costs are extremely cheap compared with lithium [6, 7]. Sodium also shows chemical properties similar to those of lithium, as both elements belong to the same group in the periodic table. The operation principle of NIB is also similar to that of LIB considering alkali ion (Na, Li ion) transports reversibly between the cathode and anode like rocking chair; however, NIB cell voltages and energy densities are much lower than those of LIBs [7,8,9]. Komaba et al. reported that the energy density of a NIB is 60% that of a LIB. Practical applications require that NIBs provide an energy density comparable to that of the LIBs. Attempts at increasing the energy density of a NIB have focused on developing electrode materials with optimal operation voltages and capacities. Because the cathode materials in a NIB are structurally similar to those in a LIB, they may be readily introduced as oxides, polyanions, or other compounds [10,11,12,13]. Graphite, a common anode material used in LIBs, however, cannot be used in NIBs because Na+ cannot intercalate into graphene interlayers in carbonate-based electrolyte due to Na+’s large radius [8, 14].
Other types of carbonaceous materials, including hard carbon, soft carbon, expanded graphite, and hollow carbon, have been investigated as possible replacements for graphite. These materials perform well electrochemically, but their capacities are limited (300 mAh g−1) and their rate capabilities are poor [14,15,16,17]. These issues may be overcome by alloying elements with Na, such as Sn, P, and Sb. These materials provide a high capacity and good rate capability that exceeds the values obtained from carbonaceous materials [18,19,20,21,22]. On the other hand, these materials suffer from the fatal drawbacks associated with a very large volume expansion (Na3P: 520%, Na3.75Sn: 490%, Na3Sb: 390%) during the reaction with Na [19]. In particular, Sb shows the relatively low-volume expansion characteristic, good electrochemical performances despite several tens of micron-meter-sized particles, and the little influence of fluoroethylene carbonate (FEC), which makes the stable solid electrolyte interface (SEI) layer on anode surface as the inevitable additive for Na alloying elements. Sb reacts favorably with group VIA elements, including O, S, and Se, to form binary compounds. Among these compounds, Sb2O3 and Sb2S3 have been tested as anode materials and provide good electrochemical performances during conversion reactions with Na [23,24,25]. Although selenium is belonged to the same group as sulfur, selenium (1 × 10−3 S m−1) has a higher electronic conductivity compared with sulfur (5 × 10−28 S m−1), which is expected to provide good electrochemical performances such as fast rate capability and cycle durability. We intend to treat a Sb–Se intermetallic compound as an anode material for NIBs. Other transition metal selenides, such as Cu2Se, MoSe2, and FeSe2, present good candidates as anode materials for use in NIBs with excellent rate capabilities and cycle performances [26,27,28]; however, the average reaction potential of 1.5 V vs Na/Na+ during the desodiation process is high, thereby reducing the cell voltage and energy density of the full cell prepared with this anode. In this study, Sb2Se3 and its composite modified with graphite were synthesized using a high-energy mechanical milling (HEMM) technique for use as the anode materials in a NIB. The Sb2Se3/C composite displayed outstanding electrochemical performances, with a high reversible capacity of 445 mAh g−1 and the average desodiation potential of 1.13 V (vs Na/Na+) at 50 mA g−1, a superior rate capability, and a very stable cycle life exceeding 200 cycles for NIB. Considering the cutoff voltage range (0.005–2.0 V vs Na/Na+) in this study, the reversible capacity, the corresponding initial Coulombic efficiency, and cyclic durability are relatively better than previous studies [29,30,31,32]. An ex situ XRD study at various reaction potentials and ex situ HR-TEM analysis of the fully sodiated and desodiated electrodes supported an electrochemical reaction mechanism in the Sb2Se3/C composite during the first cycle of sodiation/desodiation.
1.1 Experimental
Commercial Sb (99.5%, Aldrich) and Se (99.5%, Aldrich) powders were used as starting materials to synthesize Sb2Se3 and its composite. The powder mixtures were loaded and sealed under an argon atmosphere in a zirconia vial containing zirconium balls. The ball-to-powder total mass ratio was maintained at 20:1. Sb and Se powders were mechanically milled in a molar ratio of 2:3 over 12 h under an Ar atmosphere at a 300 rpm speed using a planetary mill (Pulverisette 5, Fritsch). Sb2Se3 powder was obtained using this procedure. Additionally, its composite was synthesized by an additional mechanical milling of the as-prepared Sb2Se3 (70 wt%) and an artificial graphite (MCMB, 30 wt%) over 3 h. The powder X-ray diffraction (XRD, X-Pert PRO MPD Philips) measurements were carried out under Cu Kα radiation (λ = 1.5406 Å) applied at 40 kV and 30 mA between 10° and 90° at a scan rate of 0.01°, 2θ/s. The morphology and composition of the as-prepared Sb2Se3 and its composite were determined using scanning electron microscopy (SEM, Hitachi S-4800) and transmission electron microscopy (TEM, FEI Titan G2 ChemiSTEM Cs Probe). The surface film formed on the desodiated electrode was analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Scientific, Multilab 2000).
For the electrochemical measurement, a slurry consisting of an active material (70 wt%), super P (15 wt%) as the conducting agent, and the polyacrylic acid (PAA, 15 wt%) dissolved in N-methyl-2-pyrrolidone (NMP) as a binder were pasted onto a 10 µm Cu foil, followed by drying at 120 °C under vacuum for 3 h. Coin-type cells (2032) were assembled in an Ar-filled glove box using metallic sodium as a reference and counter electrode, a 200-µm glass microporous fiber (GMF) as a separator, and 1 M NaClO4 in 1:1 ethylene carbonate (EC)/propylene carbonate (PC) (1:1, v/v, Panax Etec, Co., Ltd.) with 5% FEC as the electrolyte. The electrochemical performances were measured galvanostatically between 0.005 and 2.0 V (vs Na/Na+). All electrochemical experiments were conducted at room temperature. The capacity was calculated based on the composite weight.
2 Results and discussion
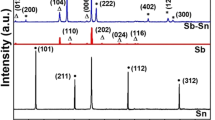
Figure 1a, b displays the XRD patterns of the as-prepared Sb2Se3 and Sb2Se3/C composite. In the Sb2Se3 powders, all peaks corresponded to crystalline Sb2Se3 (JCPDS no. 72-1184) and other crystalline phases including the starting elements (Sb, Se) were not observed. The Sb2Se3/C composite displayed a lower crystallinity than the Sb2Se3 phase, given the peak broadening as a result of the milling process involving additional HEMM with carbon. These results demonstrated that Sb2Se3 and its composite with carbon were synthesized by only HEMM with no other processes. The inset in Fig. 1 shows the crystalline structure of the Sb2Se3 compound with a space group of Pnma. Sb2Se3 crystallized in the orthorhombic lattice with a = 11.783 Å, b = 3.9799 Å, and c = 11.613 Å. Rietveld refinement of the XRD patterns obtained from Sb2Se3 revealed that the experimental pattern agreed well with the simulated results.

XRD patterns collected from a Sb2Se3 and b Sb2Se3/C (inset: crystalline structures of Sb2Se3, red: Sb and green: Se). (Color figure online)
Figure 2a, b shows bright-field and high-resolution TEM images, along with the selected-area electron diffraction (SAED) patterns, of the Sb2Se3 and Sb2Se3/C composites, respectively. Figure 2a confirms that the micron-sized Sb2Se3 particles were comprised of agglomerated highly crystalline nanocrystallites. The Sb2Se3/C composite consisted of very tiny (approximately 5–15 nm) nanocrystalline Sb2Se3 dispersed homogeneously throughout an amorphized graphite carbon matrix. The diffuse ring patterns of the Sb2Se3/C composite indicated a lower crystallinity than that of Sb2Se3. Overall, the TEM images agreed well with the XRD patterns measured from the Sb2Se3 and Sb2Se3/C composites. Energy dispersive spectroscopy (EDS) mappings of the Sb2Se3/C composite confirmed that the tiny Sb2Se3 nanocrystallites were dispersed evenly throughout the amorphous graphite carbon matrix. The Sb2Se3/C composite, therefore, was composed of nanoscale Sb2Se3 crystallites (5–15 nm) and amorphized graphite carbon. Considering that HEMM process provided an effective approach to synthesizing both Sb2Se3 and its composite.

TEM, HR-TEM images, STEM, and EDS mappings of the a Sb2Se3 and b Sb2Se3/C composites
Figure 3 shows the cyclic performances of Sb, Sb2Se3, and the Sb2Se3/C composite between 0.005 and 2.0 V (vs. Na/Na+) at 200 mA g−1. The elemental Sb electrode performed poorly during the cyclic tests, despite a high initial sodiation capacity of 590 mAh g−1. After the first several cycles, the Sb electrode underwent a dramatic capacity fading process due to the large volumetric changes resulting from Na3Sb formation [19]. This process was attributed to the pulverized active material and its corresponding electrical isolation in the electrode during the repeated sodiation and desodiation steps. Unlike Sb, elemental Se electrodes seldom undergo reversible electrochemical reactions with Na. On the other hand, the initial sodiation and desodiation capacities of Sb2Se3 were 900 and 450 mAh g−1, respectively, with an ICE of 50% (Fig. 3b). Compared with the Sb and Se electrodes, the intermetallic compounds of Sb2Se3 displayed good cycle performances with a capacity retention (398 mAh g−1) of 77.4% of the initial capacity at 40 cycles. After 40 cycles, however, the capacity decreased rapidly. Although Sb2Se3 displayed much better reversibility and cycle performances than those of the Sb or Se elemental electrodes, further improvements of cycle performance were needed. Furthermore, the ICE of Sb2Se3 was too low for commercial uses as an anode material in NIBs. On the other hand, the Sb2Se3/C composite displayed first sodiation and desodiation capacities of 575 and 408 mAh g−1 at 200 mA g−1, respectively, with a good ICE of 71%. Although the Sb2Se3/C composite displayed a lower desodiation capacity than Sb2Se3 during the first cycle, it provided a higher reversible capacity and, therefore, a 21% higher ICE compared to Sb2Se3. These results demonstrated that the Sb2Se3/C composite offered a more reversible electrochemical reaction with Na than Sb2Se3. As shown in Fig. 3d, the Sb2Se3/C composite displayed a very stable cycle durability over 200 cycles, considering that the residual capacity (401 mAh g−1) at 200 cycles corresponded to 98.2% of the initial capacity (408 mAh g−1) at 200 mA g−1 over the voltage range 0.005–2.0 V. Furthermore, the coulombic efficiency of the Sb2Se3/C composite exceeded 99.5% on average over 200 cycles, indicating that NIB full cells employing the as-prepared Sb2Se3/C composite anode with cathode material can also represent the stable cyclic performances without undergoing a significant capacity loss. The excellent electrochemical performances of the electrodes would be attributed to the homogeneously distributed 5–15 nm Sb2Se3 crystallites dispersed within the amorphous carbon conductive matrix.

Charge–discharge profiles of the a Sb, b Sb2Se3, and c Sb2Se3/C electrodes, d the cycle performances of the Sb, Sb2Se3, and Sb2Se3/C electrodes at 200 mA g−1, and e the rate capabilities of the Sb, Sb2Se3, and Sb2Se3/C electrodes
In addition to the excellent cycle stability, the Sb2Se3/C composite also exhibited an outstanding rate capability. Figure 3e shows the rate capability performances of the Sb2Se3 and Sb2Se3/C composite electrodes. These electrodes were sodiated and desodiated at current densities of 50, 200, 500, 1000, and 2000 mA g−1 over five cycles applied at each electrode. At a high current density of 2000 mA g−1 (4.5 C), with the capacity at 1 C defined as the desodiation capacity, 445 mAh g−1, at 50 mA g−1, the Sb2Se3/C composite electrodes delivered a high capacity of 329 mAh g−1, which corresponded to 74% of the capacity measured at 50 mA g−1. Meanwhile, Sb2Se3 showed a lower capacity of 157 mAh g−1 at 2000 mA g−1, which corresponded to only 30% of the desodiation capacity at 50 mA g−1. The rate capability tests were conducted at 2000 mA g−1 over 5 cycles, and the capacity of the Sb2Se3/C composite recovered to its initial capacity at 50 mA g−1. These results demonstrated that the Sb2Se3/C composite maintained the original structure of the fresh electrode during sodiation and desodiation at a high current density. The remarkably high rate capabilities indicated that the Sb2Se3/C electrode permitted Na and electrons to transfer reversibly at a high current density. These results were attributed to the high electrical conductivity of the Sb2Se3/C composite electrode with the help of a carbon-forming composite with Sb2Se3.
The morphological changes displayed by each electrode during the cycle tests were explored by collecting SEM images of the electrode surfaces. Figure 4a indicated that after 100 cycles, the Sb electrode featured a plethora of cracks on the surface that accelerated electrode degradation and yielded a poor electrochemical performance. As shown in Fig. 4b, numerous cracks also formed on the surface Sb2Se3 electrode as a result of the mechanical stresses experienced by the active materials and electrical isolation during the repeated sodiation/desodiation steps. Consequently, the morphologies of Sb2Se3 and Sb changed significantly after just 100 cycles. The electrode degradation finally resulted in poor cycle performances. On the other hand, as shown in Fig. 4c, although the surfaces coarsened and became rougher during the growth of electrode particles during the long-term reactions with Na, the Sb2Se3/C composite electrode was not fragmented after 200 cycles, and dendrites or surfaces cracks were not observed on the surface. In addition, the surface of the Sb2Se3/C composite electrode was covered with a thin and stable SEI layer that supported the long-term cycle stability.

SEM images of the three samples: a Sb fresh or, after 100 cycle, b Sb2Se3 fresh or, after 100 cycles, and c Sb2Se3/C composite fresh or, after 200 cycles
The reaction potentials were checked by plotting the differential capacity plots (DCPs) of the Sb2Se3/C composite during the first and second cycles, as shown in Fig. 5a. Three peaks during sodiation and two peaks during desodiation were observed during the first and second cycles. The positions of the sodiation peaks during the first cycle differed from those observed during the second cycle due to the irreversible reaction associated with the formation of an SEI layer during the first sodiation step. The observation of several peaks during the sodiation and desodiation steps suggested the occurrence of several electrochemical reactions. The sodiation and desodiation peaks at 0.25/0.5 and 0.73 V were attributed to the alloying/dealloying reactions of Sb with Na during the second cycle [22, 24, 25, 30]. The peak pair at 1.1/1.5 V was attributed to the alloying reaction of Na with Se in the Sb2Se3/C composite [34]. Therefore, the reversible capacity of the Sb2Se3/C composite was attributed to the conversion reactions of Sb2Se3 with Na at the each reaction potential of Sb and Se. From the second cycle to 200th cycle, the peak potentials and areas in DCP curve almost remain steady, indicating that the electrochemical reactions corresponding to sodiation and desodiation of Sb2Se3/C composite progressed reversibly. On the other hand, the desodiation peak potentials around 0.73 V moved to higher potential after 200 cycles, indicating that the desodiation resistance increased compared with previous cycles.

a Differential capacity plot of the Sb2Se3/C composite at several cycles and b its ex situ XRD patterns at first and second cycles during sodiation and desodiation
The electrochemical reactions of the amorphous Sb2Se3/C composite electrode were characterized by performing ex situ XRD analyses studies during various voltage steps in the DCP curve during the first and second cycle. After full sodiation during the first cycle, peaks characteristic of Na2Se were observed, and the peaks corresponding to Na3Sb and Sb were not observed. The XRD pattern of the fully desodiated electrode at 2.0 V displayed none of the characteristic peaks, indicating that the electrode transformed to the amorphous electrode state similar to the original one. Ex situ XRD patterns of the Sb2Se3/C composite electrode at the second cycle also showed the not significantly different features from those at the first cycle. In order to confirm the micro-structural reversibility of this electrode during sodiation/desodiation process, ex situ HR-TEM analysis of the fully sodiated and desodiated electrode was performed. As shown in Fig. 6a, both of Na3Sb and Na2Se phases were observed in the FT patterns of the fully sodiated Sb2Se3/C electrode, which indicated that Sb2Se3 phase was completely converted to Na3Sb and Na2Se phases by conversion reaction. On the other hand, only the Sb2Se3 phases of nanocrystallites around 10 nm are observed without Na3Sb, or Na2Se at fully desodiated electrode (cutoff 2.0 V, Fig. 6b), which demonstrates that fully sodiated phases of Na3Sb and Na2Se were recombined to Sb2Se3 phase during desodiation reaction. Therefore, considering the peaks position in DCP curves, ex situ XRD patterns, and ex situ HR-TEM analysis, it is confirmed that the as-prepared Sb2Se3/C electrode involved the reversible reaction electrochemically by conversion and recombination reaction.

Ex situ HR-TEM patterns of a the fully sodiated state (0.005 V vs Na/Na+) and b the fully desodiated state of Sb2Se3/C composite electrode (2.0 V vs Na/Na+)
The following reaction mechanism of the Sb2Se3/C composite can be suggested from the DCP, XRD patterns, and HR-TEM analysis of the electrode.
During the sodiation reaction:
During the desodiation reaction:
The reaction mechanisms indicated that the Sb2Se3/C composite formed reversibly during the conversion reaction with the help of the amorphized graphite carbon conductive network. This reversible transformation contributed to the outstanding electrochemical performances and cycle stability.
XPS study was conducted for investigating the influences of the surface film formed on the electrochemical performances. Figure 7 showed the Na1s and F1s spectra of Sb2Se3 and Sb2Se3/C composite electrodes at the fully desodiated states, respectively. Considering the Na1s spectra at 1072 eV and F1s spectra at 684.5 eV, the compact NaF film was supposed to be formed on the Sb2Se3/C composite electrode. On the other hand, Sb2Se3 electrode showed a lower peak intensities on both Na1s and F1s spectra, implying that Sb2Se3/C composite electrode has not compact NaF film compared with Sb2Se3/C composite electrode. Therefore, it was inferred that the stable and compact NaF, formed by the decomposition of FEC, could make the Sb2Se3/C composite electrode achieved the outstanding electrochemical performances [33, 34].

XPS spectra of as-synthesized Sb2Se3 and Sb2Se3/C composite: a Na 1s and b F 1s
3 Conclusions
In conclusion, Sb2Se3 and its carbon composite were synthesized using a simple, inexpensive, and scalable HEMM method. Sb2Se3 and its carbon composite were tested as anode materials in NIBs, revealing that the Sb2Se3/C composite displayed much better electrochemical performances, including a high initial coulombic efficiency, good cycle performance, and a better rate capability than the Sb2Se3 anode, except for the initial capacity. The Sb2Se3/C composite showed an outstanding electrochemically reversible reaction with Na corresponding to a desodiation capacity of 408 mAh g−1 with an ICE of 71% at 200 mA g−1 and a superior cycle performance of 401 mAh g−1, corresponding to 98.2% of the initial desodiation capacity at 200 mA g−1 over 200 cycles. Furthermore, at high current densities (2 A g−1 at 4.5 C), a high desodiation capacity of 329 mAh g−1, corresponding to 74% of the capacity at 50 mA g−1, was obtained, indicating good recovery to the initial capacity at 50 mA g−1. SEM images and XPS studies of the electrode surfaces confirmed that the Sb2Se3/C composite condition remained comparable to the condition of the fresh electrodes without forming cracks. An ex situ XRD and HR-TEM analysis of the Sb2Se3/C composite electrode at several potentials suggested a plausible reaction mechanism.
References
Bruce PG, Scrosati B, Tarascon JM (2008) Nanomaterials for rechargeable lithium batteries. Angew Chem Int Ed 47(16):2930–2946
Armand M, Tarascon JM (2008) Building better batteries. Nature 451:652–657
Park CM, Kim JH, Kim H, Sohn HJ (2010) Li-alloy based anode materials for Li secondary batteries. Chem Soc Rev 39:3115–3141
Dunn B, Kamath G, Tarascon JM (2012) Electrical energy storage for the grid: a battery of choices. Science 334:928–935
Grosjean C, Herrera PH, Perrin M, Poggi P (2012) Assessment of world lithium resources and consequences of their geographic distribution on the expected development of the electric vehicle industry. Renew Sustain Energy Rev 16(3):1735–1744
Hong SY, Kim Y, Park Y, Choi A, Choi NS, Lee KT (2013) Charge carriers in rechargeable batteries: Na ions vs. Li ions. Energy Environ Sci 6:2067–2081
Palomares V, Serras P, Villaluenga I, Hueso KB, Gonzalez JC, Rojo T (2012) Na-ion batteries, recent advances and present challenges to become low cost energy storage systems. Energy Envrion Sci 5:5884–5901
Komaba S, Murata W, Ishikawa T, Yabuuchi N, Ozeki T, Nakayama T, Ogata A, Gotoh K, Fujiwara K (2011) Electrochemical Na insertion and solid electrolyte interphase for hard-carbon electrodes and application to Na-ion batteries. Adv Funct Mater 21(20):3859–3867
Kim SW, Seo DH, Ma X, Ceder G, Kang K (2012) Electrode materials for rechargeable sodium-ion batteries: potential alternatives to current lithium-ion batteries. Adv Energy Mater 2(7):710–721
Yabuuchi N, Kajiyama M, Iwatate J, Nishikawa H, Hitomi S, Okuyama R, Usui R, Yamada Y, Komaba S, Nat Mater 11:512–517
Komaba S, Takei C, Nakayama T, Ogata A, Yabuuchi N (2010) Electrochemical intercalation activity of layered NaCrO2 vs. LiCrO2. Electrochem Commun 12(3):355–358
Park YU, Seo DH, Kwon HS, Kim B, Kim J, Kim H, Kim I, Yoo HI, Kang K (2013) A new high-energy cathode for a Na-ion battery with ultrahigh stability. J Am Chem Soc 135(37):13870–13878
Lee HW, Wang RT, Pasta M, Lee SW, Liu N, Cui Y (2014) Manganese hexacyanomanganate open framework as a high-capacity positive electrode material for sodium-ion batteries. Nat Commun 5:5280
Cao Y, Xiao L, Sushko ML, Wang W, Schwenzer B, Xiao J, Nie Z, Saraf LV, Yang Z, Liu Z (2012) Sodium ion insertion in hollow carbon nanowires for battery applications. Nano Lett 12(7):3783–3787
Wen Y, He K, Zhu Y, Han F, Xu Y, Matsuda I, Ishii Y, Cummings J, Wang C (2014) Expanded graphite as superior anode for sodium-ion batteries. Nat Commun 5:4033
Thomas P, Billaud D (2001) Sodium electrochemical insertion mechansims in various carbon fibres. Electrochim Acta 46(22):3359–3366
Alcantara R, Lavela P, Ortiz GF, Tirado JL (2005) Carbon microspheres obtained from resorcinol-formaldehyde as high-capacity electrodes for sodium-ion batteries. Electrochem Solid-State Lett 8(4):A222–A225
Kim Y, Park Y, Choi A, Choi NS, Kim J, Lee J, Rut J, Oh SM, Lee KT (2013) An amorphous red phosphorus/carbon composite as a promising anode material for sodium ion batteries. Adv Mater 25(22):3045–3049
Qian J, Wu X, Cao Y, Ai X, Yang H (2013) High capacity and rate capability of amorphous phosphorus for sodium ion batteries. Angew Chem Int Ed 125(17):4731–4734
Komaba S, Matsuura Y, Ishikawa T, Yabuuchi N, Murataand W, Kuze S (2012) Redox reaction of Sn-polyacrylate electrodes in aprotic Na cell. Electrochem Commun 21:65–68
Baggetto L, Ganesh P, Meisner RP, Unocic RR, Jumas J, Bridges CA, Veith GM (2013) Characterization of sodium ion electrochemical reaction with tin anodes: experimental and theory. J Power Sources 234:48–59
Darwiche A, Marino C, Sougrati MT, Fraisse B, Stievano L, Monconduit L (2012) Better cycling performances of bulk Sb in Na-ion batteries compared to Li-ion systems: an unexpected electrochemical mechanism. J Am Chem Soc 134(51):20805–20811
Hu M, Jiang Y, Sun W, Wang H, Jin C, Yan M (2014) Reversible conversion-alloying of Sb2O3 as a high-capacity, high-rate, and durable anode for sodium ion batteries. ACS Appl Mater Interfaces 6(21):19449–19455
Yu DYU, Prikhodchenko PV, Mason CW, Batabyal SK, Gun J, Sladkevich S, Medvedv AG, Lev O (2013) High-capacity antimony sulphide nanoparticle-decorated graphene composite as anode for sodium-ion batteries. Nat Commun 4:2922
Zhu Y, Nie P, Shen L, Dong S, Sheng Q, Li H, Luo H, Zhang X (2015) High rate capability and superior cycle stability of a flower-like Sb2S3 anode for high-capacity sodium ion batteries. Nanoscale 7:3309–3315
Yue JL, Sun Q, Fu ZW (2013) Cu2Se with facile synthesis as a cathode material for rechargeable sodium batteries. Chem Commun 49:5868–5870
Zhang K, Hu Z, Liu X, Tao Z, Chen J (2015) FeSe2 microspheres as a high-performance anode material for Na-ion batteries. Adv Mater 27:3305–3309
Ko YN, Choi SH, Park SB, Kang YC (2014) Hierarchical MoSe2 yolk–shell microspheres with superior Na-ion storage properties. Nanoscale 6:10511–10515
Ou X, Yang C, Xiong X, Zheng F, Pan Q, Jin C, Liu M, Huang K (2017) A new rGO-overcoated Sb2Se3 nanorods anode for Na+ battery: in-situ X-ray diffraction study on a live sodiation/desodiation process. Adv Funct Mater 27:1606242
Zhao W, Li CM (2017) Mesh-structured N-doped graphene@Sb2Se3 hybrids as an anode for large capacity sodium-ion batteries. J Colloid Interface Sci 488:356–364
Luo W, Calas A, Tang C, Li F, Zhou L, Mai L (2016) Ultralong Sb2Se3 nanowire-based free-standing membrane anode for lithium/sodium ion batteries. ACS Appl Mater Interfaces 8:35219–35226
Li W, Zhou M, Li H, Wang K, Cheng S, Jiang K (2015) Cabon-coated Sb2Se3 composite as anode material for sodium ion batteries. Electrochem Commun 60:74–77
Baggetto L, Ganesh P, Sun CN, Meisner RA, Zawodzinski TA, Veith GM (2013) Intrinsic thermodynamic and kinetic properties of Sb electrodes for Li-ion and Na-ion batteries: experiment and theory. J Mater Chem A 1:7985–7994
Bodenes L, Darwiche A, Monconduit L, Martinez H (2015) The solid electrolyte interphase a key parameter of the high performance of Sb in sodium-ion batteries: comparative X-ray photoelectron spectroscopy study of Sb/Na-ion and Sb/Li-ion batteries. J Power Sources 273:14–24
Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20152020105420). This research was supported by Technology Development Program to Solve Climate Changes through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT (No. 2018M1A2A2063343).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Choi, JH., Lee, MH., Choi, HY. et al. Investigation of electrochemical reaction mechanism for antimony selenide nanocomposite for sodium-ion battery electrodes. J Appl Electrochem 49, 207–216 (2019). https://doi.org/10.1007/s10800-018-1267-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-018-1267-2