
Abstract
Highly dispersed platinum or platinum-based catalysts on a conductive support are commonly used as electrode materials in low-temperature fuel cells. Similarly, iridium oxide is the usual anode material in polymeric exchange membrane electrolyzers. The performance and, in particular, the stability of these catalysts strongly depends on the characteristics of the support. This study presents the results of the physicochemical and electrochemical characterization of the powers of antimony-doped tin oxide (ATO) synthesized by a chemical coprecipitation method and a minimum calcination time. These supports were used as catalytic supports for oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). The ATO was characterized by X-ray diffraction (XRD), high resolution transmission electron microscopy, energy dispersive spectrometry and four probe resistivity techniques. The electrochemical properties were obtained by cyclic voltammetry (CV), linear voltammetry (LV) and rotating disk electrode (RDE). The material obtained showed nanometric sizes of 4–9 nm, and the electrochemical results indicate that the synthesized ATO nanoparticles can be used as a support for IrO2 and Pt in electrodes for PEM electrolyzers and fuel cells. Some mixtures of synthesized ATO and Vulcan carbon (VC) were assayed as mixed supports for ORR and OER and for acquiring a protective effect of ATO on the degradation of the carbon support.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
A direct chemical-electrical energy conversion process is conducted in fuel cells (FCs) with high efficiency and low pollutant emission. Highly dispersed metal or metal oxide catalysts on a conductive support are commonly used as electrode materials for oxidation and reduction reactions. An ideal support should offer the following properties: (i) good electrical conductivity; (ii) good catalyst–support interaction; (iii) large surface area; (iv) mesoporous structure enabling the ionomer and polymer electrolyte to bring the catalyst nanoparticles close to the reactants, i.e., to maximize the triple-phase boundary (TPB); (v) good water handling capability to avoid flooding; (vi) good corrosion resistance; and (vii) easy recovery of the catalyst [1, 2]. A good interaction between the catalyst and the support not only improves the catalyst efficiency and decreases catalyst loss but also leads to charge transfer. The support can also assist in sufficiently enhancing the catalyst performance and durability by reducing catalyst poisoning (e.g., CO and S), and in some cases, it also affects the catalyst particle size. Among non-carbonaceous based materials, titania, indium oxide, alumina, silica, tungsten oxide nanostructures and conducting polymers have been widely investigated with the aim of improving the issue of carbon corrosion, which is suffered in varying degrees by all carbon supports [1–3].
Tin dioxide, SnO2, belongs to the family of transition-metal dioxide compounds with rutile structures. The mineral form of SnO2 is called cassiterite, which is the main ore of tin. SnO2 is usually regarded as an oxygen-deficient n-type semiconductor. Hydrous forms of SnO2 have been described in the past as stannic acids, although such materials appear to be hydrated particles of SnO2 [4, 5]. This oxide has been proposed as a support material for fuel cell electrocatalysts because of its chemical properties: it adsorbs OH species at low potentials and/or induces the electronic effect with Pt catalysts. For its use as a catalyst support in fuel cells, however, the electrical conductance of SnO2 has to be improved. Undoped tin dioxide is a wide bandgap semiconductor (Eg ~3.6 eV) with electrical resistivity varying from 10 to 106 Ω cm, depending on the temperature and the stoichiometry of the oxide [6, 7]. Antimony is one of the most common n-type dopants for SnO2; the addition of Sb modifies the band structure of SnO2 by the donation of an extra electron into the conduction band and a substitutional replacement of a cation by the impurity dopant [8–12]. The ATO has a high tolerance to corrosion in acid media, and when it is doped with conductive species, such as Sb5+ or Sb3+, its electrical conductivity increases to 102 or 103 Ω −1 cm−1 [13–15]. Several studies indicate that the ATO could be used as a catalyst support for OER and ORR in a water electrolyzer (WE) and FCs, with a reported improvement in the stability of the support compared with an undoped oxide under the conditions of oxygen evolution [12, 13, 16–22].
In previous reports, ATO powders have mainly been synthesized using sol–gel processing, hydrothermal preparation, coprecipitation, thermal evaporation, polymer pyrolysis and solid-state reactions. The coprecipitation method from a homogeneous solution containing metallic sources is a cost-efficient procedure for electrocatalyst and support synthesis, and it presents several advantages, such as high purity, small crystalline size, short preparation time, low cost and possible large scale production. Although coprecipitation is recognized as a good method of synthesis [8, 9, 23–28], additional reproducibility, optimization and simplification studies must be performed before it can be considered to be a commercial choice for the production of ATO.
As a contribution to the field, this paper describes the results of the OER and ORR kinetics study of IrO2 and Pt catalysts supported on ATO. The support was synthesized by a simple coprecipitation method with heat treatment for 1 and 3 h. The electrochemical and physicochemical characterization of the synthesized oxide materials indicate that for using the synthesis conditions reported in this work, short calcination times are required to obtain an adequate support for the electrocatalysts of the oxygen reactions, thus decreasing the time and cost of synthesis. In this work, the results of the study of mixtures of synthesized ATO and Vulcan assayed as supports are also described; the results indicate that ATO provides a protective effect against carbon corrosion.
2 Experimental section
2.1 Synthesis of IrO2
The IrO2 used as a catalyst for OER was synthesized using a colloidal system that consisted of mixing a 0.02 M H2IrCl6·nH2O solution with 1 M KOH for 50 min at 100 °C. The dark iridium hydroxide precursor obtained was then annealed at 400 °C for 1 h [29–34].
2.2 Synthesis of antimony doped tin oxide (ATO)
ATO supports were obtained by the coprecipitation method from the reaction between SnCl4·5H2O and SbCl3 precursors (Sigma Aldrich) in a hydrochloric acid medium at moderate temperature (50 °C) for 1 h. The precipitate was dried at 90 °C for 24 h, followed by an annealing procedure at 500 °C for 1 or 3 h [35–38].
2.3 Electrodes preparation
The electrodes were prepared from a catalytic ink comprising 90 μL of Nafion® (5 wt%, Aldrich), 540 μL of spectroscopic grade ethanol (Merck), 6 mg of IrO2 or Pt (Sigma Aldrich) as catalyst and 6 mg of ATO or Vulcan as a support. Unsupported IrO2 electrodes were also prepared from a catalytic ink containing 12 mg of the active oxide and similar volumes of Nafion® and ethanol as mentioned above. The inks were obtained using a FRITSCH/PULVERISETTE 23 mini-mill for 0.5 h to homogenize the mixture and to promote particle dispersion according with the common practice to produce supported catalyst systems [39]. Later, the resulting suspensions were placed in an ultrasonic bath for 0.5 h with the aim of gaining additional dispersion of the particles in the mixture. Subsequently, a volume of 3 μL of ink was deposited on a clean polished glassy carbon disk electrode (GCE) (A = 0.07068 cm2). The coated GCEs were dried in a furnace at 80 °C for 10 min. IrO2 and Pt films supported on ATO, Vulcan carbon or their mixtures were used as working electrodes for the OER and ORR kinetic studies. All of the current values reported in this paper are normalized to the electrode geometric area.
2.4 Electrochemical characterization
All of the experiments were performed in a double-compartment electrochemical glass cell. An Hg/Hg2SO4/0.5 M H2SO4 (ESM = 0.69 V vs. NHE) reference electrode was used and positioned as close as possible to the working electrode by means of a Luggin capillary. A Pt-mesh was used as the counter-electrode, and the aqueous electrolytic medium was 0.5 M H2SO4 at room temperature. The electrochemical measurements were performed using an EG&G PAR VersaSTAT 3 Potentiostat/Galvanostat. In this study, all potential are reported with respect to the normal hydrogen electrode (NHE).
Cyclic voltammetry was conducted to evaluate the effect of different supports by scanning the potential at a rate of 50 mV s−1 between −0.16 and 1.54 V under a N2 atmosphere supporting electrolyte solution. Oxygen evolution experiments were performed by linear scan voltammetry (LSV) in the anodic direction from 0.59 to 1.69 V at a scan rate of 5 mV s−1 in an O2-free electrolyte solution at a constant electrode rotation frequency of 1000 rpm. Oxygen reduction experiments were performed using rotation speeds of 100, 225, 400, 625, 900 and 1600 rpm in a potential range of 0.3 to −0.4 V at a scan rate of 5 mVs−1 in oxygen-saturated H2SO4. The rotation rate was controlled by a PINE MSRX precision rotating system.
2.5 Physicochemical characterization
The synthesized supports were characterized by X-ray diffraction (XRD) measurements on a Bruker D8 advanced diffractometer using Cu Kα radiation (λ = 1.5406 A) within the Bragg angles 10 < 2θ < 80° in steps of 0.01°. A JEOL JEM-2200FS high resolution transmission electron microscope was used for HRTEM image characterization, and energy dispersive spectrometry (EDS) was used for the elemental analysis of the materials. Electrical Conductivity of materials was measured at room temperature by the four probes technique using an Agilent 4338B milliohmmeter. A pressure of 200 lb in−2 was applied to the powders during the conductivity measurements.
3 Results and discussion
3.1 Physicochemical characterization
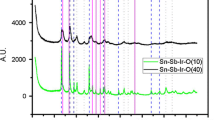
The XRD diffraction patterns of ATO are shown in Fig. 1. The characteristic peaks of SnO2 with a cassiterite tetragonal structure and a rutile phase are observed. Due to the minimal dopant quantity used, any diffraction peak related to Sb could not be identified. The diffractograms correspond to ATO at 1 h (black) and 3 h (red) annealing times (Fig. 1). A characteristic SnO signal peak can be identified at 29 degrees in the red diffractogram. Some other peaks assigned to the SnO2 are also detectable, but the characteristic 110 and 101 peaks are overlapped with the SnO 112 peak.

X-ray diffraction patterns of the ATO obtained by coprecipitation and calcination at 500 °C
Figure 2a shows the SEM micrograph of 4 wt% antimony-doped tin oxide nanoparticle calcined for 1 h. The surface morphology of the ATO powder showed the presence of irregular particles ranging from 60 to 70 nm. The TEM micrograph shown in Fig. 2b reveals that the particles of ATO are composed of an agglomeration of small homogeneous crystallites with sizes ranging from 4 to 9 nm. The synthesized material was nanocrystalline in nature with an estimated average size of 6 nm, as is shown in the histogram of Fig. 2c, which corresponds to a low dispersion polycrystalline powder [37, 40–42]. The particle sizes are smaller than the reported average value of 7–20 nm for commercial ATO [15, 43].

a SEM micrograph of ATO powder, b TEM micrograph, c histogram of the particle sizes (N = 1000)
Figure 3 shows a HR-TEM image and a typical electron diffraction pattern of an ATO 1 h nanocrystalline grain. The electron diffraction (ED) rings can be indexed to the pattern of ATO with a cassiterite structure. The small size of the ATO particles is responsible for the somewhat thicker diffraction circles in Fig. 3b. The typical reported lattice parameters of SnO2 changed when the Sn ions were replaced by Sb ions. The ionic radii of Sn4+, Sb5+ and Sb3+ are 0.72, 0.62 and 0.90 Å, respectively, and when Sb5+ enters the SnO2 structure, the lattice parameters became smaller. By contrast, if Sb3+ ions replace the Sn4+ ions, an increase in the lattice parameters is observed [9, 10, 23, 44].

a HR-TEM micrographs of ATO and b the electron diffraction pattern
The calculated lattice parameters from the diffraction rings (Fig. 3b) are shown in Table 1. We can see that the obtained parameters for the ATO powders were slightly different than the standard values for undoped tin oxide. The decrease in lattice parameters can be attributed to the substitution of Sn4+ ions by Sb5+ ions in the crystal structure of ATO during the synthesis step. This result is in good agreement with those reported in previous literature [9], which also confirm that Sb5+ is the dominant species at low doping levels of antimony.
From the lattice-fringes analysis of HR-TEM images for the synthesized ATO (Fig. 4), the particles were composed of regularly ordered crystallites with a lattice distance of 3.34 Å corresponding to the (d 110) inter- planar spacing. This value was slightly smaller than 3.458 Å, which corresponds to the standard lattice spacing of the SnO2 sample (JCPDS PDF# 41-1445). This result confirms the formation of a tetragonal crystal structure of Sb-doped SnO2 [44, 45].

HR-TEM image of ATO
The abundance histograms of atomic Sn and Sb were obtained from the linear scanning EDS spectra of ATO shown in the SEM micrograph of Fig. 5a. According to this analysis, Tin (red line) is more abundant than antimony (green line), Fig. 5b, c, respectively. The metallic elements content in the EDS spectra of the different sample regions is shown in Table 2. In the synthesis step, a precursors mixture was placed in the reactor such that the theoretical amount of Sb dopant was 16 wt% with respect to the total metals weight, corresponding to an atomic ratio of Sn: Sb = 6:1. However, the EDS analysis indicated that only between 2 to 4 wt% of dopant is present in the final material. This means that the average atomic ratio of Sn:Sb is 17:1 which corresponds to a compound with the approximate formula: SnSb0.06O1.94, assuming that the rest of the atoms were oxygen. Nevertheless, additional research is required to verify the chemical composition and discard a phase mix.

EDS atomic abundance histograms of ATO, Sn (red line) and Sb (green line). (Color figure online)
The electronic conductivity (σ) of different material supports found in the literature and the measured electrical conductivity of the synthesized ATO supports in this work are listed in Table 3. One can note that the conductivity of the tin oxide is greatly improved by adding antimony as a dopant. The measured σ values for the ATO supports were higher that the ATO material previously reported in [12]. Nonetheless, the measured values, 2 and 1.08 S cm−1, were lower by one order of magnitude than the conductivities of some substoichiometric titanium oxides reported in the literature [45, 46].
3.2 Electrochemical characterization
3.2.1 Evaluation of the supports by cyclic voltammetry
Previous studies on the relationship between the annealing time and the electrochemical performance of ATO showed that a short time of heat treatment results in a better performance [16, 46]. In a previous report, it was shown that only 3 h of calcination are required to obtain an adequate ATO material as a support for IrO2 electrocatalysts. For instance, an OER onset potential on IrO2 supported on ATO was measured to be very similar to that observed when Vulcan carbon was used as support [48]. Therefore, in this work, only 1 h and 3 h annealing times were tested.
Figures 6a, b show typical cyclic and linear scan voltammograms, respectively, for the electrodes prepared with unsupported and supported IrO2 catalysts in a N2 saturated 0.5 M H2SO4 solution. The CV diagram for the unsupported IrO2 (Fig. 6a, green line) shows the typical pseudo-capacitive behavior, with a broad wave which could be attributed to the overlapped quasi-reversible redox couple peaks of the Ir3+/Ir4+ and Ir4+/Ir5+ transitions at ~0.68 and ~1.2 V, respectively, in acidic media. Additionally, the electrochemical response for IrO2 was almost the same with the use of any two different ATO supports. The current density amplitude are equivalent in both voltammograms which can be considered to be an evidence of a similar dispersion of IrO2 on both supports. The off-peak potential for oxygen evolution on the IrO2 unsupported electrode appears near 1.45 V while for the two IrO2/ATO electrodes appears near 1.5 V.

a Cyclic voltammogram and b linear scan voltammetry, of GCEs coated with IrO2/ATO and IrO2 in N2 saturated 0.5 M H2SO4 and room temperature at 50 mV s−1. IrO2 (solid line), ATO 1 h (dotted line) and ATO 3 h (dashed line). LV normalized to the weight of the catalyst
The electrocatalytic response respect to the OER is more clearly appreciated in the linear voltammograms (Fig. 6b). In this figure, the current density was normalized to the weight of IrO2 catalyst on the electrode area. One can see that on the IrO2 unsupported electrode the OER begins at a less anodic potential compared to that observed for the IrO2 supported on either of the two ATOs. The higher slope of the voltammograms response for the IrO2 supported on ATO makes that at a potential near to 1.6 V the curve for the unsupported IrO2 electrode intersects indicating that the catalytic activity of supported catalyst is then higher than that of the IrO2 electrodes at potentials >1.6 V. Furthermore, the amount of IrO2 in the unsupported electrodes is twice as much as that used in the supported electrodes. Thus, it means that a similar current density is achieved with the half amount of IrO2 catalyst in the supported case. This is a clear evidence that the synthesized ATO material behaves as an effective catalyst support for the OER. The lack of electroactivity of the ATO by itself towards the OER (data not shown) indicates that does not exist a co-catalytic or synergetic effect. Finally, the voltammograms for the iridium oxide supported on ATO (1 h) and ATO (3 h) are similar (Fig. 6b), thus, an equivalence in the electrochemical response of the two ATO materials as a support at a different annealing time (1 or 3 h) are indistinct for the IrO2 electroactivity. According to this result, only ATO at 1 h was considered for further electrochemical characterization in this work.
The suitable electrochemical behavior of ATO as a support for OER electrocatalyst was already known [12, 15, 35, 38, 46, 48–51], and this was also confirmed with the results presented in this work. However, its use as a support for ORR has not been thoroughly studied. The trend in the development of supports and catalysts is to optimize the synthesis of such materials. In this context, there is a search for new materials that could be used interchangeably as supports for the OER and ORR. A main application of these dual materials may be in the development of unitized regenerative fuel cells (URFC). Thus, in this work, additional experiments were performed to evaluate the catalytic activity of Pt and IrO2 supported on mixtures of ATO and Vulcan carbon (VC) for the oxygen electrochemical reactions. The proportions of supports and catalysts used in the preparation of the different electrodes are presented in Table 4.
Figure 7 compares the cyclic voltammograms obtained for Pt, Fig. 7a, and IrO2, Fig. 7b, supported on mixtures of different proportions of Vulcan and ATO in a N2 saturated 0.5 M H2SO4 solution. The characteristic peaks related with the electrochemical surface processes on Pt are attenuated when the carbon proportion in the mixture is decreased. Nevertheless, the typical electrochemical behavior of platinum in acidic medium is still outlined in all voltammograms. Similarly, in the voltammograms corresponding to the IrO2 catalyst there are broader waves corresponding to the redox reactions over the Ir surface when Vulcan is in a higher proportion in the support as shown in Fig. 7b. These results are consistent with the higher electronic conductivity of carbon with respect to that of ATO leading to a higher surface charge. In Fig. 7, the potential onset for oxygen evolution on the Pt electrodes appears near 1.7 V, whereas the corresponding oxygen evolution peaks on IrO2 electrodes appear at a 300 mV lower anodic potential, confirming then the high electrocatalytic properties of iridium oxide for OER. In addition, it can be noted that in the mixed supports, their relative composition has a minimal effect on the overpotential required for oxygen evolution according to the CV curves.

Cyclic voltammograms of a Pt and b IrO2 supported on 25 % ATO + 75 % CV (dotted line), 50 % ATO + 50 % CV (dashed line), 75 % ATO + 25 % VC (solid line), in N2 saturated 0.5 M H2SO4 at 50 mV s−1
More detailed information of the electrochemical behavior for the mixed support electrodes could be obtained from the linear voltammetry measurements. Figure 8a shows the j–E diagrams obtained for ORR using the RDE technique at a constant rotating speed of 1800 rpm. There is a difference in the limiting currents for Pt electrodes on different mixed supports that may be attributed to the surface coverage of the Pt catalyst and to the physical properties, such as the porosity and nature of the catalyst/support material film on the glassy carbon electrode. The RDE curves show the classical mixed, kinetic and diffusion control zones; although these regions are better defined when the Vulcan is in a large amount in the support mixtures. On the other hand, Fig. 8b shows the linear scan voltammograms for the OER of IrO2 on different mixed supports; the voltammograms remain practically unchanged regardless of the composition of the mixed support. In all cases, the OER begins at a potential near to 1.54 V, although higher current densities are obtained when a greater ATO proportion is used in the mixed support.

RDE for ORR of Pt a and linear scan voltammograms for OER of IrO2 supports on 25 % ATO + 75 % VC (dotted line), 50 % ATO + 50 % VC (dashed line), and 75 % ATO + 25 % VC (solid line) in solution of 0.5 M H2SO4 at v = 5 mV s−1
Figure 9 shows the results of the chronoamperometric stability determination for IrO2 electrodes on mixed supports at potential conditions where the OER is significant. This figure also shows the behavior of IrO2 supported on Vulcan carbon only (open circles). The chronoamperograms were obtained at an applied potential of 1.63 V for 8 h. During the test period, the electrodes of IrO2 supported on the Vulcan and ATO mixtures showed a slight loss in current density compared with that exhibited by electrodes prepared with only carbon. Although the initial current density of the Vulcan electrode was the highest, this electrode lost its catalytic activity after 6 h, whereas on the mixed support electrodes, the oxygen evolution remains constant for more than 8 h. Even in the case of a mixed support with a higher proportion of Vulcan carbon (75 wt%), there was an improvement in the corrosion tolerance with respect to the carbon by itself as a support. These results indicate that ATO provides protection to Vulcan as compared to the usual degradation observed for carbonaceous materials at high operational potentials commonly employed in electrolyzer systems.

Eight hour chronoamperograms for the OER of IrO2 supported on 25 % ATO + 75 % VC (dotted line), 50 % ATO + 50 % VC (dashed line), 75 % ATO + 25 % VC (solid line) and 100 % VC (times line) in 0.5 M H2SO4 at 1.63 V and room temperature
Further evidence of the stability of the supports could be obtained from the OER and then the ORR consecutive performance using mixed Pt and IrO2 catalysts and mixed ATO and VC supports with the equal proportions described in item 7 of Table 4. Figure 10 shows the RDE and LSV plots obtained from 100 sequential experiments between the ORR and OER conditions of a rotating electrode. Figure 10a shows the RDE plots for the ORR obtained from the first to the 100th measurement (for simplicity, only four curves of the series are presented). An important difference in the reduction density current was for the OER between sequential experiments. A decrease in the OER current densities was observed between the first and last voltammograms, as shown in Fig. 10b. This gradual loss in activity for the OER can be attributed to an IrO2 wearing from the catalytic mixture due to the rotation of the electrode. However, these sequential experiment results indicate that the synthesized ATO + VC mixed material can be used equally as a support for both electrochemical oxygen reactions.

Sequential consecutive curves: a RDE curves for ORR and b LSV curves for OER of mixed Pt + IrO2 catalyst supported on mixed ATO + VC electrodes in O2 saturated 0.5 M H2SO4. 1st (dashed line), 25th (dotted line), 50th (open square), 75th (times) and 100th (solid line) sequential voltammograms. ν = 5 mVs−1
The Tafel plots for oxygen reactions on pure and 50:50 wt% mixed ATO and VC supports are shown in Fig. 11. The Tafel plots for the OER were obtained from LSV curves after ohmic drop (IRs) correction due to the electrolyte resistance (Rs). The Rs values were obtained using electrochemical impedance spectroscopy (EIS) [52]. The Tafel plots for ORR were obtained from RDE curves after correction by the diffusion current contribution. Figure 11a shows the Tafel plots for the ORR of Pt supported on the three supports. The lines are nearly coincident and fit to slopes near 120 mV dec−1, suggesting that the reduction reaction is conducted on the Pt surface with the transfer of the first electron as the determinant step of the reaction. On the other hand, the three Tafel lines for the OER on IrO2 on the different supports corresponded to a slope value near 60 mV dec−1, Fig. 11b. This value has been observed for the OER on several metal oxides in the low overpotential region (η low), corresponding to a reaction-limiting step related to the reorganization of oxygen-species over the catalyst surface before the transfer of the first electron. At values of log j > 1.6 V, Tafel slopes could be obtained with higher values near 120 mV dec−1 (η high), reflecting a change in the rate determining step of the mechanism. This behavior has also been observed for metal oxide electrodes [53, 54].

Tafel plots for a ORR on Pt and b OER on IrO2, supported on ATO (open triangle), VC (times), and 50:50 wt%. ATO + VC (open rhombs) in 0.5 M H2SO4 and room temperature
A summary of the particular Tafel slopes (b) obtained for oxygen reactions on different electrodes along with other kinetic parameters, such as the mass transfer coefficient (α) and exchange current (j o ) values, are given in Tables 5 and 6 for the ORR and OER, respectively.
From the results presented in Table 5, it can be argued that the similar values obtained in j o and j at 0.8 V indicate that the change of support had a minimal effect on the ORR kinetic parameters. The support played a more important role in the OER, as it can be conclude from the different values reported in Table 6. The onset potential, and the exchange and relative current densities of oxygen generation diverge when the support is changed. However, the α and b values for both electrochemical oxygen reactions confirm that the mechanisms of ORR and OER are independent of the support and depend only on the catalyst. The results also indicate that OER is faster than ORR on the supports and under the experimental conditions studied.
4 Conclusions
Nanoparticles of antimony-doped tin oxide were obtained from a simple coprecipitation synthesis method. Only short times of calcination (1 or 3 h) were required to obtain a material that had properties as a support for IrO2 and Pt catalysts. The synthesized ATO showed quasi-spherical shaped particles of an average size of 6 nm, with a Sb dopant composition in the range of 2–4 wt%. The electrochemical and kinetics results presented in this work indicate that the synthesized ATO can be used indistinctly in the ORR and OER. When ATO was used in mixtures with Vulcan carbon, the mixed support showed an improved electrochemical stability for OER at a potential near 1.6 V with respect to those observed for a carbon only support. The materials reported here have potential applications in PEM fuel cells, water electrolyzers and unitized regenerative fuel cells.
References
Antolini E (2010) Composite materials: an emerging class of fuel cell catalyst supports. Appl Catal B 100:413–426
Sharma S, Pollet BG (2012) Support materials for PEMFC and DMFC electrocatalysts—a review. J Power Sources 208:96–119
Long NV, Nogami M et al (2013) The development of mixture, alloy, and core-shell nanocatalysts with nanomaterial supports for energy conversion in low-temperature fuel cells. Nano Energy 2:636–676
Oviedo J, Gillan MJ (2000) Energetics and structure of stoichiometric SnO2 surfaces studied by first-principles calculations. Surf Sci 463:93–101
Rajendran V, Anandan K (2012) Size, morphology and optical properties of SnO2 nanoparticles synthesized by facile surfactant-assisted solvothermal processing. Mater Sci Semicond Process 15:393–400
Antolini E, Gonzalez ER (2009) Ceramic materials as supports for low-temperature fuel cell catalysts. Solid State Ion 180:746–763
Zhang D, Tao L et al (2006) Surface morphologies and properties of pure and antimony-doped tin oxide films derived by sol–gel dip-coating processing. Mater Chem Phys 100:275–280
Lu HF, Hong RY et al (2012) Preparation of ATO nanorods and electrical resistivity analysis. Mater Lett 68:237–239
Krishnakumar T, Jayaprakash R et al (2009) Structural, optical and electrical characterization of antimony-substituted tin oxide nanoparticles. J Phys Chem Solids 70:993–999
Hu P, Yang H, Ouyang J (2012) Synthesis and characterization of Sb–SnO2/kaolinites nanoparticles. Appl Clay Sci 55:151–157
Hu Y, Zhang H, Yang H (2008) Synthesis and electrical property of antimony-doped tin oxide powders with barite matrix. J Alloy Compd 453:292–297
Xu J, Qingfeng L et al (2012) Antimony doped tin oxides and their composites with tin pyrophosphates as catalyst supports for oxygen evolution reaction in proton exchange membrane water electrolysis. Int J Hydrog Energy 37:18629–18640
Wang LS, Lu HF et al (2012) Synthesis and electrical resistivity analysis of ATO-coated talc. Powder Technol 224:124–128
Zhukova AA, Rumyantseva MN et al (2009) Influence of antimony doping on structure and conductivity of tin oxide whiskers. Thin Solid Films 518:1359–1362
Marshall AT, Haverkamp RG (2010) Electrocatalytic activity of IrO2–RuO2 supported on Sb-doped SnO2 nanoparticles. Electrochim Acta 55:1978–1984
Cruz JC, Rivas S, Beltran D et al (2012) Synthesis and evaluation of ATO as a support for Pt–IrO2 in a unitized regenerative fuel cell. Int J Hydrog Energy 37:13522–13528
Jung DW, Park DW (2009) Synthesis of nano-sized antimony-doped tin oxide (ATO) particles using a DC arc plasma jet. Appl Surf Sci 255:5409–5413
Kim DW, Kim DS et al (2006) Preparation of hard agglomerates free and weakly agglomerated antimony doped tin oxide (ATO) nanoparticles by coprecipitation reaction in methanol reaction medium. Mater Chem Phys 97:452–457
Wu X, Scott K (2011) RuO2 supported on Sb-doped SnO2 nanoparticles for polymer electrolyte membrane water electrolysers. Int J Hydrog Energy 36:5806–5810
Gurrola MP, Guerra M et al (2013) High surface electrochemical support based on Sb-doped SnO2. J Power Sources 243:826–830
Pan C, Li Y et al (2011) Platinum–antimony doped tin oxide nanoparticles supported on carbon black as anode catalysts for direct methanol fuel cells. J Power Sources 196:6228–6231
You DJ, Kwon K et al (2009) Platinum–antimony tin oxide nanoparticle as cathode catalyst for direct methanol fuel cell. Catal Today 146:15–19
Sladkevich S, Kyi N et al (2011) Antimony doped tin oxide coating of muscovite clays by the Pechini route. Thin Solid Films 520:152–158
Liu TJ, Jin ZG et al (2008) Conducting antimony-doped tin oxide films derived from stannous oxalate by aqueous sol–gel method. Appl Surf Sci 254:6547–6553
Zhang D, Deng Z et al (2006) Microstructure and electrical properties of antimony-doped tin oxide thin film deposited by sol–gel process. Mater Chem Phys 98:353–357
Benrabah B, Bouaza A et al (2011) Impedance studies of Sb doped SnO2 thin film prepared by sol gel process. Superlattices Microstruct 50:591–600
Yang F, Zhang XJ et al (2007) Preparation of highly dispersed antimony-doped tin oxide nano-powder via ion-exchange hydrolysis of SnCl4 and SbCl3 and azeotropic drying. Chin J Aeronaut 20:181–186
Yang F, Zhang XJ et al (2007) Preparation of highly dispersed antimony-doped tin oxide nanopowders by azeotropic drying with isoamyl acetate. Trans Nonferrous Metals Soc China 17:626–632
Cruz JC, Bablio V et al (2012) Nanosized Pt/IrO2 electrocatalyst prepared by modified polyol method for application as dual function oxygen electrode in unitized regenerative fuel cells. Int J Hydrog Energy 37:5508–5517
Siracusano S, Baglio V et al (2010) Electrochemical characterization of single cell and short stack PEM electrolyzers based on a nanosized IrO2 anode electrocatalyst. Int J Hydrog Energy 35:5558–5568
Vazquez GL, Ferro S et al (2006) Preparation and characterization of RuO2–IrO2–SnO2 ternary mixtures for advanced electrochemical technology. Appl Catal B 67:34–40
Zhang Y, Wang Ch et al (2007) Deposited RuO2–IrO2/Pt electrocatalyst for the regenerative fuel cell. Int J Hydrog Energy 32:400–404
Mayousse E, Maillard F et al (2011) Synthesis and characterization of electrocatalysts for the oxygen evolution in PEM water electrolysis. Int J Hydrog Energy 36:10474–10481
Papazisi KM, Siokou A et al (2012) Preparation and characterization of IrxPt1−xO2 anode electrocatalysts for the oxygen evolution reaction. Int J Hydrog Energy 37:16642–16648
Hu P, Yang H (2010) Controlled coating of antimony-doped tin oxide nanoparticles on kaolinite particles. Appl Clay Sci 48:368–374
Yang H, Hu Y, Qiu G (2002) Preparation of antimony-doped SnO2 nanocrystallites. Mater Res Bull 37(15):2453–2458
Zheng M, Wang B (2009) One-step synthesis of antimony-doped tin dioxide nanocrystallites and their property. Trans Nonferrous Metals Soc China 19:404–409
Xiaozhen L, Beiling Q (2007) Complexation-coprecipitation synthesis and characterization of erbium and antimony doped SnO2 conductive nanoparticles. J Rare Earths 25:72–76
Li N, Meng Q, Zhang N (2014) Dispersion stabilization of antimony-doped tin oxide (ATO) nanoparticles used for energy-efficient glass coating. Particuology 17:49–53
Bai F, Yun He et al (2006) One-step synthesis of monodispersed antimony-doped tin oxide suspension. Mater Lett 60:3126–3129
Liu G, Xu J et al (2014) Nanosphere-structured composites consisting of Cs-substituted phosphotungstates and antimony doped tin oxides as catalyst supports for proton exchange membrane liquid water electrolysis. Int J Hydrog Energy 39:1914–1923
Zhang J, Gao L (2004) Synthesis and characterization of antimony-doped tin oxide (ATO) nanoparticles by a new hydrothermal method. Mater Chem Phys 87:10–13
Gurrola MP, Gutiérrez J et al (2014) Evaluation of the corrosion of Sb-doped SnO2 supports for electrolysis systems. Int J Hydrog Energy 39:16763–16770
Du Y, Yan J et al (2012) Fabrication and excellent conductive performance of antimony-doped tin oxide-coated diatomite with porous structure. Mater Chem Phys 133:907–912
Chen X (2005) Synthesis and characterization of ATO/SiO2 nanocomposite coating obtained by sol–gel method. Mater Lett 59:1239–1242
Siracusano S, Stassi A et al (2013) Preparation and characterisation of Ti oxide based catalyst supports for low temperature fuel cells. Int Lorunal od Hydrog Energy 38:11600–11608
Siracusano S, Baglio V et al (2009) Preparation and characterization of titanium suboxides as conductive supports of IrO2 electrocatalysts for application in SPE electrolysers. Electrochem Acta 54:6292–6299
Ávila VV, Cruz JC (2013) Electrochemical Study of Sb-Doped SnO2 Supports on the Oxygen Evolution Reaction: Effect of Synthesis Annealing Time. Int J Electrochem Sci 8:10586–10600
Jung D, Park D (2009) Synthesis of nano-sized antimony-doped tin oxide (ATO) particles using a DC arc plasma jet. Appl Surf Sci 255:5409
Manesse M, Sanjines R et al (2008) Preparation and characterization of antimony-doped SnO2 thin films on gold and silver substrates for electrochemical and surface plasmon resonance studies. Electrochem Commun 10:1041–1043
Yin M, Junyuan X et al (2014) Highly active and stable Pt electrocatalysts promoted by antimony-doped SnO2 supports for oxygen reduction reactions. Appl Catal B 144:112–120
Tsuchiva H, Kobayashi O (2004) Mass Production cost of PEM fuel cell by learning curve. Int J Hydrog Energy 29:985–990
Cruz JC, Baglio VS et al (2011) Preparation and characterization of RuO2 catalysts for oxygen evolution in soild polymer electrolyte. Int J Electrochem Sci 6:6607–6619
Escalante IL, Durón SM et al (2010) Electrochemica characterization of IrO2-Pt and RuO2-Pt mixtures as bifunctional electrodes for unitized regenerative fuell cell. J New Mater Electrochem Syst 13:227–233
Acknowledgments
The authors wish to thank the Mexican CONACyT (Project 167012) for financial support of this work. V. Ávila-Vázquez thanks CONACyT for a graduate fellowship. In addition, we would like to thank MSc. Carlos Ornelas for his help in the HR-TEM sample characterization at the Laboratorio Nacional de Nanotecnología, CIMAV.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ávila-Vázquez, V., Galván-Valencia, M., Ledesma-García, J. et al. Electrochemical performance of a Sb-doped SnO2 support synthesized by coprecipitation for oxygen reactions. J Appl Electrochem 45, 1175–1185 (2015). https://doi.org/10.1007/s10800-015-0876-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-015-0876-2