
Abstract
Mesoporous nickel cobaltite (NiCo2O4) nanoparticles were synthesized via a hydrothermal and soft-templating method through quasi-reverse-micelle mechanism. The physicochemical properties of the NiCo2O4 materials were characterized via X-ray diffraction, scanning electron microscopy, X-ray photoelectron spectra, and nitrogen sorption isotherms measurements. The electrochemical performances of the NiCo2O4 electrode were investigated by cyclic voltammetry, chronopotentiometry, and electrochemical impedance spectroscopy tests. The obtained NiCo2O4 materials exhibit typical mesoporous structures, with an average particle size of about 200 nm, a specific surface area of 88.63 m2 g−1, and a total pore volume of 0.337 cm3 g−1. The facile electrolytes penetration for the mesoporous structures favors high-performance of the NiCo2O4 electrode. The NiCo2O4 electrode shows a high specific capacitance (591 F g−1 at 1 A g−1), high-rate capability (248 F g−1 at 20 A g−1), and a good cycling behavior for tested 3,000 cycles, indicating a promising application for electrochemical capacitors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Environmental pollution, climate change, and exhaust of fossil fuels have triggered the ever-growing worldwide interest in developing sustainable and clean energy products [1, 2]. Electrochemical capacitors (ECs), as an important next generation of energy storage device, are now attracting intensive attention, mainly due to their higher power density than batteries and higher energy density than conventional dielectric capacitors [2, 3]. This property makes ECs ideal to augment traditional batteries in many different applications. However, to become primary devices for practical power supply, ECs must be developed further to improve their specific capacitance, rate capability, cycling stability, and operational safety [4].
As for high-performance ECs, it is key step to fabricate advanced electrode materials with the clean, efficient, and versatile use of energy. It has been reported that spinel nickel cobaltite (NiCo2O4) offers many intriguing advantages of high electrochemical activity, low-cost, abundant resources, and environmentally friendly [5, 6]. Therefore, NiCo2O4 material has been expected to be a promising cost-effective and scalable alternative to the state-of-art ECs material ruthenium dioxide (RuO2) [7, 8]. To our knowledge, NiCo2O4 has become the focus candidate for ECs since the early report by Wei et al. [6].
It is noted that particle properties, such as morphology, size, porosity, etc., have a profound effect on the electrochemical performance of the materials, which is highly related with the route of material design and specific preparation method. In recent years, mesoporous materials (e.g., mesoporous carbon or metal oxides) have been extensively investigated for ECs applications because of their excellent supercapacitive performance [9]. Among various mesoporous materials, mesoporous NiCo2O4 nanostructures [6, 10–13] have attracted particular interests and attentions due to their large specific capacitance, high-rate capability, and excellent cycling stability.
Soft-templating (ST) method is regarded as an effective technique to fabricate mesoporous materials, especially for mesoporous metal oxides, through the oil–water interface self-assembly processes between organic phase and inorganic precursors in certain solvents by applying various template agents including surfactants such as sodium dodecyl sulfate (SDS), cetyltrimethylammonium bromide (CTAB), dodecyl amine (DDA), etc. or Pluronics block copolymers like poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO, with different forms of P123, F127, etc.). In addition, hydrothermal technique is a desirable method to fabricate functional nanomaterials for ECs due to its high temperature and pressure environments of particle growth. Therefore, combining these two techniques can be expected to obtain advanced materials for high-performance ECs.
In this work, nanostructured mesoporous NiCo2O4 materials were synthesized via a hydrothermal and ST combined method, in which SDS is chosen as the template. The physicochemical properties and electrochemical performances of the NiCo2O4 materials were characterized and investigated, respectively. The results demonstrate that the synthesized mesoporous NiCo2O4 nanomaterials exhibit high specific capacitance, high-rate performance, and good cycling behavior, indicating a promising candidate for high-performance ECs.
2 Experimental
2.1 Synthesis of materials
All the reagents used in the experiment were of analytical grade and were used without further purification. In a typical procedure, Co(NO3)2·6H2O (0.6667 g), Ni(NO3)2·6H2O (0.3333 g), and SDS (1.0000 g) were dissolved in deionized water (20 mL) under continuous magnetic stirring at 40 °C in a water bath for 30 min to form a homogenous solution. Then, NaOH (0.3 M, 22 mL) aqueous solution was poured into the above-generated solution with continuous magnetic stirring at 40 °C for another 30 min to form a well-mixed solution. Subsequently, the resulting solution was transferred into a Teflon-lined stainless-steel autoclave (50 mL) and heated at 140 °C for 12 h in a box oven, and next cooled naturally to the room temperature. The precipitates were collected by centrifugal filtration, washed with distilled water and absolute ethanol several times, afterward dried at 60 °C for 12 h under vacuum condition, and finally annealed at 300 °C for 3 h in air atmosphere at a heating rate of 1 °C min−1 in a muffle stove to obtain the ultimate products. The material synthesis processes via a quasi-reverse-micelle mechanism (template self-assembly process) are illustrated in Scheme 1. The SDS (C12H25SO4Na) is a typical anionic surfactant. First, the C12H25 \({\text{SO}}_{4}^{ - }\) anions strongly absorb the hydrated Ni2+ and Ni2+ cations as a result of electrostatic interactions, thus the quasi-reverse-micelles can be formed through the interface self-assembly processes between the soft template and inorganic precursors. Subsequently, the added OH− anions continuously react with the hydrated Co2+ and Ni2+ cations in the reverse-micelles to form the –OH–Ni–OH–Co–OH– networks. And then, the nickel–cobalt hydroxides/SDS composite nanostructures were formed under the hydrothermal process. Finally, the SDS templates were removed by the washing and calcinations and meanwhile the mesoporous structures were generated among the NiCo2O4 nanoparticles.

Schematic representation of the mesoporous NiCo2O4 nanoparticles formation process: a schematic representation of a quasi-reverse-micelle, and b mesoporous NiCo2O4 formation process
2.2 Material characterizations
Thermogravimetry (TG) and differential thermal analysis (DTA) were carried out with a STA 449 F3 Jupiter thermogravimetric analyzer (NETZSCH) in the temperature range of 40–810 °C in air at a heating rate of 10 °C min−1. X-ray diffraction (XRD) patterns were recorded on a Rigaku-Dmax 2500 diffractometer equipped with graphite monochromatized Cu Kα radiation source (λ = 1.5406 Å) at a scanning speed of 4° min−1 in the 2θ range of 10–80°. Scanning electron microscopy (SEM) images were conducted using Philips XL 30 and JEOL JSM-6700F microscopes. X-ray photoelectron spectra (XPS) were measured using an ESCALAB-MKII spectrometer (UK) with Al Kα radiation (1,486.6 eV) as the X-ray source for excitation, the raw spectra were curve-fitted by nonlinear least squares fittings with a Gauss–Lorentz ratio (80:20) using the XPSPEAK41 software. Surface area and porosity were determined by nitrogen sorption isotherms at 77 K using a Micromeritics ASAP 2020 Analyzer, prior to analysis, the sample was degassed under vacuum at 120 °C for 24 h. The specific surface area (SSA) was calculated using the multipoint Brunauer–Emmett–Teller (BET) method, while the pore size distribution (PSD) and pore volume data were calculated from the desorption branches of nitrogen sorption isotherms based on the Barrett–Joyner–Halenda (BJH) equation.
2.3 Electrochemical performances investigations
The electrochemical performances of NiCo2O4 electrode were examined by cyclic voltammetry (CV), chronopotentiometry (CP), and electrochemical impedance spectroscopy (EIS) tests, which were carried out in a conventional three-electrode electrochemical setup containing a NiCo2O4 working electrode (WE), a platinum plate (3 cm × 4 cm) counter electrode (CE), and a Hg/HgO (1 M KOH, aqueous) reference electrode (RE). The WE was fabricated by pressing the homogenized mixture of 66.7 wt% active material (as-synthesized NiCo2O4), 33.3 wt% conductive agent and binder (teflonized acetylene black, TAB; the mass ratio of AB and PTFF is 2:1) on the current collector (stainless steel mesh, 1 cm2 area), and followed by drying in a vacuum oven at 80 °C for 12 h. The mass loading of active materials was about 4 mg cm−2.
CV tests were cycled between the voltage of 0 and 0.53 V at the scan rate of 2 mV s−1, CP tests were cycled between the voltage of 0 and 0.5 V with a current density range from 1 to 20 A g−1, EIS experiments were taken under a constant direct current (dc) bias potential of open circuit voltage (OCV) with an alternating current (ac) amplitude of 5 mV over the sinusoidal alternating voltage frequency range of 100 kHz–10 mHz. Cycle life test was conducted via a CP measurement between the voltage of 0 and 0.5 V at the current density of 5 A g−1 for consecutive 3,000 cycles. The electrolytes used for tests were 6 M KOH aqueous solutions. All tests were carried out at room temperature (about 20 °C).
The gravimetric specific capacitance (C m, F g−1) and coulombic efficiency (η, %) measured from CP curves were calculated according to Eqs. (1) and (2), respectively.
where m (g) represents the mass of active materials, i (A g−1) is current density, ΔV (V) is the applied potential range, Δt d and Δt c are the time of discharging and charging, respectively.
3 Results and discussions
3.1 Characterizations of the NiCo2O4 materials
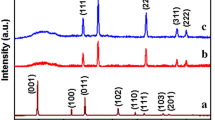
Figure 1 shows the XRD patterns of as-prepared precursors. As shown in the figure, the precursors are mainly composed of two kinds of crystalline phases of Co(OH)2 (JCPDS No. 30-0443) and Ni(OH)2 (JCPDS No. 74-2075) with the high crystallinity. Meanwhile, some diffraction peaks of the CoCo2O4 crystalline phase (JCPDS No. 80-1543) and Ni2O3H crystalline phase (JCPDS No. 40-1179) were detected, which is resulted from the partial oxidations of the Co(OH)2 and Ni(OH)2 phases in the existence of \({{\text{NO}}_{3}^{ - }}\) anions under the high temperature and pressure circumstances of the hydrothermal processes. In addition, a spot of SDS signals (JCPDS No. 04-0006) were also observed, indicating the remains of the template in the precursors.

XRD patterns of as-prepared precursors
TG–DTA measurements were conducted to investigate the thermal behavior of the precursor in air. As illustrated in Fig. 2a, there are three distinct weight loss steps in TG plots, corresponding to two times changes in the direction of heat flow of DTA curves which are shown in Fig. 2b. The first step (3.4 % weight loss) occurs in the temperature range between 40 and 150 °C with one DTA endothermic peak, corresponding to the evaporation of physical adsorbed water. The second step (28.6 % weight loss) occurs in the temperature range between 150 and 400 °C with two continuous but different DTA exothermic peaks (the former is short and weak, the latter is sharp and strong), which is probably caused by the difference of reaction rates during conversion process of precursor. Here, the two exothermic peaks associate with combustion of remains of surfactant template and thermal decomposition of the precursor powders to formation of NiCo2O4 spinels [14]. Note that the removal of physisorbed water and surfactant remains will open up more pore spaces and lead to larger pore volume and pore size, which favors the enhancement of porosity of the materials. The third step (8.4 % weight loss) occurs in the temperature range from 400 to 800 °C with a broad DTA endothermic peak, owing to decomposition of the NiCo2O4 spinels [15]. On the basis of the decomposition temperature range (150–400 °C) and the peak temperature value (270 °C), the temperature for calcinations was set at 300 °C for 3 h to ensure a complete decomposition of the precursor.

a TG and b DTA curves of as-prepared precursors
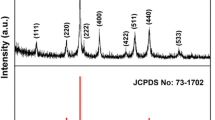
Figure 3 shows the XRD patterns of the NiCo2O4 materials. The resultant diffraction peaks given in the figure corroborate with the standard patterns for cubic NiCo2O4 (JCPDS No. 73-1702) with spinel structure. The sharp and well-defined diffraction lines illustrate high crystallinity of the materials. The inset of Fig. 3 shows a picture of the NiCo2O4 spinel unit cell, the lattice parameter a 0 of the materials determined from the observed d-spacing for crystal plane (311) at 2θ value of 36.8° by using Eqs. (3) and (4), for a cubic lattice, is 0.8104 nm, basically corresponding with the standard value of 0.8110 nm for spinel NiCo2O4 (JCPDS No. 20-0781).
where λ is the wavelength of X-ray radiation (1.5406 Å), β is the full width at half maximum of the diffracted peak, θ is the Bragg angle of diffraction, d is observed space for crystal plane, h, k, l are the Miller indices.

XRD patterns of as-synthesized NiCo2O4 materials
The SEM images of the NiCo2O4 materials are shown in Fig. 4a–d. The observed primary NiCo2O4 particles exhibit a hexagonal morphology with diameter of ca. 200 nm. Meanwhile, there are some light aggregations which may be formed during the calcinations process, because the primary particles might sinter each other and grow into large secondary particles when calcinations [16]. However, the obtained materials still show a porous structure and this can provide easy access for ions to access the electrode/electrolyte interface, which is a very important factor for the surface Faradaic reactions.

SEM images of as-synthesized NiCo2O4 materials with different scale bars a 1 μm, b 500 nm, c and d 200 nm
The nitrogen adsorption–desorption isotherms, corresponding PSD and pore volume plots of the NiCo2O4 materials are shown in Fig. 5. The nitrogen sorption isotherms can be classified as type IV with a H3-type hysteresis loop according to the IUPAC classification, typical mesoporous materials. The BET SSA, BJH cumulative pore volume, and average pore diameter of the materials are 88.63 m2 g−1, 0.337 cm3 g−1, and 18.8 nm, respectively. Moreover, pore size exhibits a bimodal size distribution at 7.8 and 17.5 nm due to expansion of pores, indicating high porosity of the NiCo2O4 materials. Here, the not so high SSA is mainly attributed to the large particle size (200 nm), and the light agglomerations of particles may cause some loss of pore volumes.

a Nitrogen adsorption/desorption isotherms, b pore size distribution plots, and c pore volume plots of as-synthesized NiCo2O4 materials
The chemical bonding states and compositions of surface elements of as-synthesized NiCo2O4 materials were evaluated by XPS which are shown in Fig. 6. It is apparent that typical signals of O1s, Co2p, and Ni2p core levels are detected in the survey scan spectra shown in Fig. 6a. Of which, the O1s spectra, as presented in Fig. 6b, show four oxygen contributions, denoted as O1 (529.3 eV), O2 (530.3 eV), O3 (531.5 eV), and O4 (532.8 eV), associating with the typical of metal–oxygen bond [17, 18], the oxygen in hydroxyl groups [19], the high number of defect sites with low-oxygen coordination in the material with small particle size [20], and the multiplicity of physi-/chemi-sorbed water at and within the surface [17, 18], respectively. The Co2p spectra shown in Fig. 6c consist of two spin–orbit doublets characteristics of Co2+ and Co3+ and two shakeup satellites (identified as “Sat.”). Similarly, the Ni2p spectra given in Fig. 6d are composed of two spin–orbit doublets characteristic of Ni2+ and Ni3+ and two shakeup satellites. These results show that the surface of as-synthesized NiCo2O4 materials has a composition containing Co2+, Co3+, Ni2+, and Ni3+ species. Therefore, the formula of NiCo2O4 can be generally expressed as follows: \({\text{Co}}_{1 - x}^{2 + } {\text{Co}}_{x}^{3 + } [{\text{Co}}^{3 + } {\text{Ni}}_{x}^{2 + } {\text{Ni}}_{1 - x}^{3 + } ]{\text{O}}_{4}\) (0 < x < 1) (the cations within the square bracket are in octahedral sites and the outside ones occupy the tetrahedral sites) [21, 22]. Here, the surface quantitative analysis of the NiCo2O4 materials based on the XPS were also conducted, which displays that the surface O/Co/Ni atomic ratio of the materials is 5.16/1.08/1, which should be 4/2/1 for the stoichiometric NiCo2O4, indicating a much enrichment in Ni species and the heavy surface non-stoichiometry of the materials, which further validates the fact that the intensity of typical of metal–oxygen bond (O1, 17.7 %) is much weaker than the sum of the oxygen in hydroxyl groups bond (O2, 17.4 %) and the high number of defect sites with low-oxygen coordination bond (O3, 51.4 %) in the O1s spectra (Fig. 6b). While the some higher O atom ratio may be resulted from the absorption of physi-/chemi-sorbed water due to the capillary condensation and high surface activity of nanostructural porous NiCo2O4 materials, which corresponds to the presence of the multiplicity of physi-/chemi-sorbed water bond (O4, 13.5 %) in the O1s spectra (Fig. 6b).

XPS profiles of as-synthesized NiCo2O4 materials: a survey scan, b O1s core levels, c Co2p core levels, and d Ni2p core levels
3.2 Investigations of the NiCo2O4 electrode
CV is a very useful tool to determine various electrode processes, electrode reaction mechanisms, electrochemical reversibility, dynamic parameters, etc. Figure 7a shows the CV plots at 2 mV s−1, the whole CV plots can be divided into three electrode processes: (i) a minor electric double-layer (EDL) process (0–0.1 V), (ii) a primary charge-transfer process (0.1–0.5 V) with one pair of broad and strong redox peaks marked as (A, C), which originates from the overall contributions of redox couples Co3+/Co2+ and Ni3+/Ni2+ [23, 24], (iii) a small sharp area of irreversible electrocatalytic oxygen evolution reaction process (0.5–0.53 V). Here, the electrode process is obviously dominated by the redox peaks, while the EDL occupies the much minor position, indicating the dominant Faradaic behavior of the NiCo2O4 electrode. Hence, the overall capacitance contributions of the NiCo2O4 electrode come from the minor EDL capacitance part and the major Faradaic capacity part which could be expressed as the Eq. (5) [25].
where NiCo2O4∥OH− represents the EDL formed by the hydroxyl ion (OH−), and NiCo2O4–OH represents the product formed by oxidation reaction involving the hydroxyl ion.

a CV plots of as-fabricated NiCo2O4 electrode at a scan rate of 2 mV s−1, b CP curves of as-fabricated NiCo2O4 electrode at different current densities (1–20 A g−1), c specific capacitance and capacitance retention of as-fabricated NiCo2O4 electrode as a function of the current density, and d cycling behavior of as-fabricated NiCo2O4 electrode at a current density of 5 A g−1
Figure 7b shows the CP curves at 1–20 A g−1. The charging/discharging branches present lightly asymmetric images, indicating quasi-reversible dynamics feature. Furthermore, the distinct nonlinear CP curves and obvious charging/discharging plateaus validate the dominant Faradaic behavior of the electrode, corresponding well with CV discussions. The specific capacitance and corresponding retentions under different current densities (1–20 A g−1) are depicted in Fig. 7c, which exhibits a considerable specific capacitance range of 591–248 F g−1 and a high retention domain of 100–42 % when the current density increases from 1 to 20 A g−1, demonstrating the high capacitance and rate capability. The reasons for the merits of the mesoporous NiCo2O4 electrode may be that mesopores are capable of overcoming the primary kinetic limits of electrochemical processes [26] and make the penetration of electrolytes into the whole oxide matrix facilely and further reduce the diffusion resistance, which means that mesoporous microstructures are believed to facilitate larger capacitance and higher power rates [27].
Long cycling life is a very important factor for the electrode materials used in ECs. Figure 7d shows the cycling behavior of the NiCo2O4 electrode at a high-rate of 5 A g−1. As demonstrated in the figure, the capacitance went through a vertical decay in the initial several cycles and then gradually decreased in the initial 100 cycles, with an obvious specific capacitance decay from 480 to 312 F g−1, which is mainly resulted from the loss of irreversible energy of the NiCo2O4 electrode, i.e., the irreversible surface intercalation/extraction redox reactions in the lattice-embedded NiCo2O4 spinels. The deeper reason may be due to the heavy surface non-stoichiometry property of Co, Ni valent atoms of the materials (the intensity of metal–oxygen bond O1 (17.7 %) is much lower than the sum (68.8 %) of the high number of defect sites with low-oxygen coordination bond O3 and the oxygen in hydroxyl groups bond O2), resulting in the surface structural unstability and the irreversible surface intercalation/extraction redox reactions. Similar phenomenon has been also reported for the TiO2 electrode for ECs with the LiClO4 aqueous electrolytes [28], which capacity dropped sharply before 200 cycles and tended to be stable after 500 cycles, and the reason is that the percentage of surface metal–oxygen bond Ti–O–Ti (7.6 %) (Ti4+) is much lower than the surface metal–hydroxyl groups bond Ti–OH (70.0 %) (Ti3+) after 500 cycles, which resulted in the irreversible lithium intercalation reactions and the capacity reduction. After the initial 100 cycles, however, the NiCo2O4 electrode reached a much more stable state and showed a high cycling stability in the next 2,900 cycles, exhibiting 90 % retention (281 F g−1) based on the value (312 F g−1) of the 100th cycle. The coulombic efficiency of the electrode during all cycling periods (except the initial several cycles) maintained nearly at 100 %, showing a high coulombic efficiency under a high-rate condition. Based on the above analysis, the NiCo2O4 electrode exhibits a good cycling behavior, implying a promising application for the ECs.
To further investigate the electrochemical behavior of the NiCo2O4 electrode, EIS tests were conducted before and after 3,000 cycles. Figure 8 shows the Nyquist plots of experimental impedance data and fitted results of the NiCo2O4 electrode. All the measured impedance spectra are similar in shape with an arc in a higher frequency region and a spike in a lower frequency region. The measured impedance spectra were analyzed using the complex nonlinear least-squares (CNLSs) fitting method [29] on the basis of the equivalent circuit, which is given in the inset of Fig. 8. The model circuit consists of five elements: the equivalent series resistance (R e), the constant phase element (CPE) (Q), the charge transfer resistance (R ct), the Warburg impedance (Z W), and the limit capacitance (C l). The R e is a combinational resistance of ionic resistance of electrolyte, intrinsic resistance of active material, and contact resistance at the electrode/current collector interface [30]. The R ct, the semicircle in the high-frequency range caused by the Faradaic reactions, represents the kinetic resistance to charge-transfer at electrode/electrolyte boundary or intrinsic charge-transfer resistance of porous electrode. The Z W, the slope of the approximate 45° portion of the Nyquist plots in the mid-frequency range, is a result of the frequency dependence of ion diffusion/transport in the electrolyte to the bulk of electrode [31]. C l is the limit capacitance [29]. The CPE (Q) is used in place of double-layer capacitance (C dl) at the electrode/electrolyte boundary. The impedance of CPE is defined as Eq. (6):
where Y 0 is the frequency-independent constant relating to the surface electroactive properties, w is the radial frequency, the exponent n arises from the slope of log Z versus log f (and has values −1 ≤ n ≤ 1). If n = 0, the CPE behaves as a pure resistor; n = 1, CPE behaves as a pure capacitor, n = −1, CPE behaves as an inductor; while n = 0.5 corresponds to Warburg impedance (Z W) which is associated with the domain of mass transport control arising from the diffusion of ions to and from the electrode/electrolytes interface. The obtained values of R e, C dl, R ct, Z W, and C l were calculated from CNLS fitting of the experimental impedance spectra and presented in Table 1. The R e, C dl, and C l values before and after 3,000 cycles are almost the same, while a major difference comes from the R ct and Z W. After 3,000 cycles, the calculated charge-transfer resistance for the electrode is increased from 6.535 to 27.40 Ω, which is probably due to the intrinsic quasi-reversible dynamics feature of the electrode that can cause the continuous accumulations of charge-transfer resistance during the repeated charge/discharge cycling under a high-rate condition and the inevitable electrode structural deteriorations after high-rate long-term charge/discharge cycling (such as the loss of adhesion of some active materials with the current collector or the over-oxidation of current collector) that make against charge-transfer reactions. The value of Warburg resistance parameter after 3,000 cycles decreased from 0.1303 to 0.07888, which is probably due to the more generated bulk Faradaic reactions in the NiCo2O4 electrode that delayed the fast electrolyte ions diffusion and migration after the long-term charge/discharge cycles.

Nyquist plots of experimental impedance data and fitted results of as-fabricated NiCo2O4 electrode in fresh and after 3,000 cycles (inset shows the equivalent circuit model)
4 Conclusions
A hydrothermal and soft-templating method was exploited to fabricate mesoporous NiCo2O4 nanomaterials. The obtained NiCo2O4 materials display an average particle size of about 200 nm, a specific surface area of 88.63 m2 g−1 and a total pore volume of 0.337 cm3 g−1, exhibiting typical mesoporous structures. The NiCo2O4 electrode shows a high specific capacitance (591 F g−1 at 1 A g−1), high-rate capability (248 F g−1 at 20 A g−1), and a good cycling behavior for tested 3,000 cycles, indicating a promising application for ECs.
References
Aricò AS, Bruce P, Scrosati B, Tarascon JM, Van Schalkwijk W (2005) Nat Mater 4:366
Liu C, Li F, Ma LP, Cheng HM (2010) Adv Mater 22:E28
Simon P, Gogotsi Y (2008) Nat Mater 7:845
Burke A (2000) J Power Sources 91:37
Cui B, Lin H, Li JB, Li X, Yang J, Tao J (2008) Adv Funct Mater 18:1440
Wei TY, Chen CH, Chien HC, Lu SY, Hu CC (2010) Adv Mater 22:347
Wang HL, Gao QM, Jiang L (2011) Small 7:2454
Wei TY, Chen CH, Chien HC, Lu SY, Hu CC (2009) Adv Mater 21:1
Liu Y, Zhao WW, Zhang XG (2008) Electrochim Acta 53:3296
Yuan CZ, Hou LR, Zhang XG, Shen LF, (David) Lou XW (2012) Adv Funct Mater 22:4592
Zhang GQ, (David) Lou XW (2013) Adv Mater 25:976
Zhang GQ, (David) Lou XW (2013) Sci Rep 3:1470
Zhang GQ, Wu HB, Hoster HE, Chan-Park MB, (David) Lou XW (2012) Energy Environ Sci 5:9453
Chi B, Li JB, Han YS, Chen YJ (2004) Int J Hydrog Energy 29:605
Peshev P, Toshev A, Gyurov G (1989) Mater Res Bull 24:33
Kim H, Kim YH, Joo JB, Ko JW, Yi JH (2009) Microporous Mesoporous Mater 122:283
Choudhury T, Saied SO, Sullivan JL, Abbot AM (1989) J Phys D 22:1185
Marco JF, Gancedo JR, Gracia M (2000) J Solid State Chem 153:74
Roginskaya YE, Morozova OV, Lubnin EN, Ulitina YE, Lopukhova GV, Trasatti S (1997) Langmuir 13:4621
Jimenez VM, Fernandez A, Espinos JP, Gonzalez-Elipe AR (1995) J Electron Spectrosc Relat Phenom 71:61
Kim JG, Pugmire DL, Battaglia D, Langell MA (2000) Appl Surf Sci 165:70
Zhong JH, Wang AL, Li GR, Wang JW, Ou YN, Tong YX (2012) J Mater Chem 22:5656
Wei TY, Chen CH, Chang KH, Lu SY, Hu CC (2009) Chem Mater 21:3228
Ding R, Qi L, Jia MJ, Wang HY (2012) J Appl Electrochem 42:1033
Choi D, Blomgren GE, Kumta PN (2006) Adv Mater 18:1178
Wang DW, Li F, Liu M, Lu GQ, Cheng HM (2008) Angew Chem Int Ed 47:373
Chang KH, Hu CC, Chou CY (2007) Chem Mater 19:2112
Yin J, Qi L, Wang HY (2011) ACS Appl Mater Interfaces 3:4315
Fabio AD, Giorgi A, Mastragostino M, Soavi F (2001) J Electrochem Soc 148:A845
Gamby J, Taberna PL, Simon P, Fauvarque JF, Chesneau M (2001) J Power Sources 101:109
Stoller MD, Park SJ, Zhu YW, An JH, Ruoff RS (2008) Nano Lett 8:3498
Acknowledgments
We gratefully acknowledge the financial support of this research by National Basic Research Program of China (2012CB932800, 2011CB935702), Scientific Research Foundation for the Returned Overseas Chinese Scholars and State Education Ministry (SRF for ROCS and SEM), and Hundred Talents Program of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ding, R., Qi, L., Jia, M. et al. Hydrothermal and soft-templating synthesis of mesoporous NiCo2O4 nanomaterials for high-performance electrochemical capacitors. J Appl Electrochem 43, 903–910 (2013). https://doi.org/10.1007/s10800-013-0580-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-013-0580-z