
Abstract
Solid alumina was reduced by electro-deoxidation to aluminium metal containing 1.8 and 5.4 at% Ca in molten CaCl2–NaCl and CaCl2–LiCl electrolytes at 900 °C, respectively. The potential-pO2− diagrams for the Al–O–M–Cl (M = Na or Li, or/and Ca) system were constructed to predict equilibrium phase relationships in the electrolytes at 700 and 900 °C. It was found that calcium aluminates were formed as the main intermediate reaction products and were subsequently reduced to form the Al-rich Al–Ca alloys during electro-deoxidation. Calcium and/or lithium, at reduced activities, were created at the cathode especially at 700 °C at the same time as the ionization of the oxygen from the cathode, which resulted in Al2Ca formation. The experimental results were consistent with the thermodynamic predictions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Aluminium and its alloys are widely used as structural and functional materials in transportation, packing, construction, electrical, and many other advanced applications. Primary Al is commercially produced by the Hall–Heroult process [1, 2]. The basic features have remained essentially unchanged since the process was developed more than 100 years ago, although significant optimization of process variables has taken place to cut production costs and reduce greenhouse gas emissions [3–6].
Possibilities of using alternate Al reduction processes have been investigated extensively over several decades to reduce overall production costs and to achieve environmental benefits. The alternative processes studied so far include [1, 2, 7, 8]: (i) carbothermic reduction of Al2O3 to Al, (ii) direct reduction of bauxite to Al–Si alloys, (iii) Bayer/Al2O3–C-anode in chloride electrolytes, (iv) electrolysis of aluminous ores/alloy separation and purification, (v) Bayer/carbochlorination/bipolar cell, (vi) carbochlorination of ore/separation and purification of metal chlorides/bipolar cell, and (vii) Bayer/carbonitridation or carbosulfidation/electrolysis of AlN or Al2S3.
In this paper, we describe an electro-deoxidation method and report a set of conditions under which the oxygen is electrochemically removed from solid Al2O3 or in situ formed aluminates by making the oxides the cathode in a bath of molten CaCl2-based chlorides. The method of electro-deoxidation has been successfully used to produce many metals and alloys from their oxide precursors using the CaCl2- and LiCl-based electrolytes [9–23]. In this work, solid Al2O3 acted as the cathode in molten CaCl2-alkali chloride electrolytes. When potentials imposed to the cathode were more cathodic than oxygen ionization potentials of the Al2O3 or aluminates, but less cathodic than those needed for cathodic deposition of Ca or alkalis from the chloride electrolytes, the cathodic ionization of the oxygen from the oxides occurred through Reaction 1 rather than the cathodic deposition of the cations from the electrolytes due to Reaction 2:
where [O] is the O in a solid Al2O3 or aluminate, Mn+ and M are the metallic cation and the metal, respectively, and n is the number of electrons transferred. The ionized oxygen (O2−) dissolved into the electrolyte, diffused from the cathode to the anode where it discharged, and the metal was left behind at the cathode. The anodic reactions with the carbon anode are given by
The overall cell reaction, by combining Reaction 1 with 3, may be simply as follows:
2 Experimental
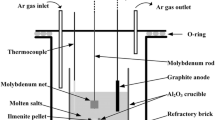
Fully dense Al2O3 tubes, of two different sizes (O.D. = 8 mm and I.D. = 5 mm; O.D. = 6 mm and I.D. = 4 mm, 99.8%, Ceramic Oxides Fabricators) were used as the starting material for Al2O3 cathodes. CaCl2 · 2H2O (greater than 99%, Aldrich) and anhydrous LiCl (99%, Sigma) were dehydrated in air at 150 °C for 1 h, heated at a rate of 6 °C h−1 to 350 °C, and then held at 350 °C for 12 h. A certain quantity of CaO may be generated during the drying process. The Li-saturated LiCl–Li melt was prepared by in situ electrolysis of molten LiCl at 700 °C until the LiCl–Li melt saturated with Li was produced. NaCl (at least 99.5%, Fluka) was dried in air at 120–150 °C for at least 5 h prior to use. Nb sheet (0.127 mm thick, 99.5%, Fansteel Inc.) was employed to fabricate a Nb basket for containing cathode materials. Figure 1 shows a schematic diagram of the electrolytic cell used in this work. High density graphite rods (Tokai Carbon Grade G347, 500 × 6 mmØ, Carbon Products of Australia; 100 × 10 mmØ, Graphite Technologies) were used as the anodes, respectively. An Al2O3 tube (O.D. = 10 mm and I.D. = 6.1 mm, 99.8%, Ceramic Oxides Fabricators) was used as the sheath for a certain section of the graphite rod anode. A stainless steel crucible was used to contain the molten salt electrolyte. Kanthal® wire (1 mmØ, Kanthal Australia Pty Ltd) and Ni wire (1.62 mmØ, Austral Wright Metals) were used as the electrode leads, respectively. The cell was placed inside a sealed Inconel reactor, which was continuously purged with dried high-purity argon throughout the experiments. The moisture and metallic impurities in the electrolytes were removed, while the quantities of CaO and Li2O in the salts were very low and both of the oxides were also electrochemically decomposed to a great extent by pre-electrolysis between the graphite rod anode and the stainless steel crucible at a cell voltage of 2.8 V under the argon at 900 °C for 10 h or longer.

Schematic diagram of the fused salt electrolytic cell used in this work (A. Kanthal wire, B. Ni wire, C. carbon anode, D. Al2O3 sheath, E. stainless steel crucible, F. Al2O3 tube, G. Nb basket, and H. fused salt electrolyte)
Electro-deoxidation was carried out at controlled potentials of 3.1 V at 900 °C and 3.4 V at 700 °C between the oxide cathode and the graphite rod anode, with a potential of 2.8 V being imposed between the crucible auxiliary cathode and the same carbon anode to prevent corrosion of the crucible. After electrolysis, the Nb basket was lifted to the top chamber of the reactor, allowing the cathode products to be argon-quenched and the salts remaining in the products to be solidified, thus preventing oxidation. The basket containing the cathode products was then taken out of the reactor at room temperature and was washed extensively in cold distilled water, followed by acetone rinse.
The cathode products were characterized using an optical microscope, a scanning electron microscope (SEM) with an energy dispersive spectrum (EDS) and energy dispersive X-ray (EDX) analysis, inductively coupled plasma (ICP), and X-ray diffractometry (XRD).
3 Results and discussion
A CaCl2–NaCl melt containing 32.2 mol% NaCl and an equimolar CaCl2–LiCl mixture were used as the electrolytes for electro-deoxidation of solid Al2O3. CaCl2 was chosen as the major component of the electrolytes due to its several desirable properties: very high solubility for O2− (e.g., 20 mol% CaO in CaCl2 at 900 °C [24]), extremely high solubility of CaCl2 in water (1.59 g mL−1 of H2O), low cost, very low toxicity, and ready availability. The addition of NaCl or LiCl to molten CaCl2 can lower liquidus temperatures and increase electrical conductivities of the electrolytes [25].
3.1 Thermodynamic considerations
Thermodynamic properties in the Al–O–M–Cl (M = Na or Li, or/and Ca) system at 700 and 900 °C are represented using the predominance diagrams on which the processes of electro-deoxidation are critically evaluated. The method of constructing the diagrams from pertinent thermodynamic data was based on those described by Littlewood [26], where the equilibrium potential E (relative to the standard chlorine electrode, S.Cl.E.) is plotted against the activity of O2− expressed as its negative logarithm, pO2−, in the chloride melts for Al. The E-pO2− diagrams summarise chemical and electrochemical properties of Al in the electrolytes at a given temperature as a function of pO2−, from which equilibrium phase relationships in the oxide/metal/salt systems can be better understood.
Ferry et al. used the E-pO2− diagrams of Fe–Ti–O compounds in molten LiCl–KCl eutectic at 470 °C to predict operating conditions for selective chlorination of TiO2 and subsequent electrowinning of metallic Ti in the melts [27]. The E-pO2− diagram for the Ti–O system at 550 °C was recently made by Martinez et al. to study the chemical and electrochemical properties of solutions of Ti chlorides in an equimolar CaCl2–NaCl mixture [28]. More recently, Inman et al. constructed the predominance diagrams for studying the direct electrochemical reduction of solid TiO2 and mixed oxide precursors in molten CaCl2 at around 900 °C [29–31]. Yasuda et al. utilized the E-pO2− diagram of Si in molten CaCl2 at 850 °C to study the direct electrolytic reduction of SiO2 [32]. The E-pO2− diagrams of Al in molten LiCl–KCl eutectic at 470 °C have been constructed by Picard et al. to investigate the chlorination of Al2O3 in molten salts [33–35]. In their diagrams, complex oxides of Al and Li were excluded. Little has been reported so far on the E-pO2− diagrams of Al in CaCl2–NaCl and CaCl2–LiCl melts at 700 and 900 °C, including various aluminates.
Construction of E-pO2− diagrams requires the definitions of standard states for the species involved. For the present systems, they are described as follows: (i) pure solid invariant compounds have unit activities, (ii) CaO at unit activity is denoted as an O2− activity of one, (iii) pure O2 or Cl2 gas at 1 atm is taken as its standard state, (iv) Ca or alkalies at unit activity is regarded as its standard state, and (v) metal chloride at unit activity is chosen as the standard state for its cation, e.g., pure CaCl2 is selected as the standard state for Ca2+. In the following E-pO2− diagrams, diagonal dot lines are iso-pressure lines for O2 (\( {\text{p}}_{{{\text{O}}_{ 2} }} \)) in the gaseous phase in equilibrium with chloride melt, while horizontal dot lines are iso-activity lines where \( {\text{a}}_{\text{Ca}} \) and \( {\text{a}}_{\text{Li}} \) are the activities of Ca and Li dissolved in their chlorides, respectively, and \( {\text{a}}_{\text{Ca}} \) [Al2Ca(s)], \( {\text{a}}_{\text{Ca}} \) [Al4Ca(s)], and \( {\text{a}}_{\text{Li}} \) [AlLi(s)] are the activities of Ca and Li in molten CaCl2 and LiCl in equilibrium with Al2Ca(s), Al4Ca(s) and AlLi(s), respectively.
3.1.1 Combined E-pO2− diagrams of Al in molten CaCl2 and a CaCl2–NaCl melt containing 32.2 mol% NaCl at 700 and 900 °C
CaO is soluble in molten CaCl2-based salts, e.g., 18 mol% CaO in molten CaCl2 and 15 mol% CaO in molten CaCl2–NaCl mixture containing 32.2 mol% NaCl at 850 °C [24, 36]. However, solubilities of Na2O and CaO in molten NaCl are negligibly small [37, 38]. An inspection of the Al2O3–CaO phase diagram shows that Al2O3 and CaO form a series of Ca aluminates in the Al2O3-rich region [39]. Ca aluminates are likely to form through chemical reactions between solid Al2O3 and CaO in CaCl2-based electrolytes and are thus considered in the construction of the E-pO2− diagrams. There are no available activity data for NaCl and CaCl2 in molten CaCl2–NaCl solutions at 700 and 900 °C in the literature. However, Chartrand and Pelton reported that molten CaCl2–NaCl solutions exhibit negative deviations from ideality at 825 and 850 °C, respectively, and changes in temperature have little influence on NaCl activities over the entire composition range [40]. It is therefore reasonable to assume that there is no temperature dependence of NaCl activity in the solutions between 700 and 900 °C. It is further assumed that molten CaCl2–NaCl solutions behave regularly. Activities of CaCl2 and NaCl in the CaCl2–NaCl melt containing 32.2 mol% NaCl at 700–900 °C are thus calculated to be 0.582 and 0.163, respectively, using the relevant activity data [40]. Decomposition potentials of the present CaCl2–NaCl melt can be then calculated from the calculated activities at 700 and 900 °C, respectively.
Figure 2a, b shows combined E-pO2− diagrams of Al in molten CaCl2 (dashed line) and a melt of CaCl2–NaCl containing 32.2 mol% NaCl (solid line) at 700 and 900 °C, respectively, as constructed using thermodynamic data [33, 40, 41]. At both temperatures, the addition of NaCl to molten CaCl2 slightly widens the electrochemical window owing to decreasing CaCl2 activity, but this effect is likely to be offset by cathodic deposition of Ca–Na alloys. As can be seen from Fig. 2, chemical interactions between Al2O3 and dissolved CaO to produce Ca aluminates require higher activities of O2−, i.e., lower pO2− values, in the CaCl2–NaCl compared to molten CaCl2. It can also be seen that solid Al2O3 or Ca aluminates are likely to reduce to metallic Al and Al2Ca or Al2Ca alone at less cathodic potentials in the CaCl2–NaCl melt than molten CaCl2, depending on cathodic potential and O2− activity. The formation of Al2Ca is more favourable than metallic Al over low pO2− regions. As an example, both Al2O3 and Ca aluminates can be reduced to form Al2Ca only at a cathodic potential of 3.1 V versus S.Cl.E. over the pO2− region of 0.3–8.5 at 700 °C or 0–6.1 at 900 °C in the CaCl2–NaCl melt. Furthermore, it is important to note that all oxides become more stable as temperature decreases. For instance, Ca12Al14O33 is even thermodynamically more stable than the CaCl2–NaCl melt at pO2− values less than 0.3 at 700 °C. Similarly, pO2− regions where Ca12Al14O33 is more stable than the electrolytes under other conditions can also be read from Fig. 2.

Combined potential-pO2− diagrams of Al in molten CaCl2 and a CaCl2–NaCl melt containing 32.2 mol% NaCl at a 700 °C and b 900 °C
3.1.2 Combined E-pO2− diagrams of Al in molten CaCl2 and a LiCl melt at 700 and 900 °C
Li2O has appreciable solubilities in molten LiCl, e.g., 16 mol% Li2O in molten LiCl at 750 °C [42], whereas solubilities of CaO in molten LiCl are very low, e.g., 0.44–0.96 mol% CaO at 700–900 °C [38]. Wang et al. reported that the solubility of CaO in an equimolar CaCl2–LiCl melt is 13.32 mol% CaO at 750 °C [43]. Hence, Li2O dissolved in molten LiCl or an equimolar CaCl2–LiCl melt is likely to react with solid Al2O3 to produce Li aluminates, as predicted from the Al2O3–Li2O phase diagram [39]. Due to the lack of thermodynamic data for LiAl5O8, this phase is thus excluded in E-pO2− diagrams of Al in molten LiCl. Figures 3a and b show combined E-pO2− diagrams of Al in molten CaCl2 (solid line) and LiCl (dashed line) at 700 and 900 °C, respectively. It can be seen from Fig. 3 that Al2O3 is more stable in molten LiCl than CaCl2 and thermodynamic stabilities of LiAlO2 are much higher than Ca aluminates at 700 or 900 °C. The stabilities for all the oxides increase, but the activities of Ca in the melt required to form solid Al2Ca become much smaller with decreasing temperature. It can be further seen that LiAlO2 is thermodynamically more stable than molten LiCl when the values of pO2− are less than 2.2 at 700 °C and 1.8 at 900 °C.

Combined potential-pO2− diagrams of Al in molten CaCl2 and a LiCl melt at a 700 °C and b 900 °C
From a thermodynamic viewpoint, solid Al2O3 and Ca aluminates may be electro-deoxidized to metallic Al and/or Al2Ca over certain regions of pO2− values at imposed cell voltages of 3.4 V at 700 °C and 3.1 V at 900 °C. Cl− ions from a chloride electrolyte are unlikely to be discharged at the carbon anode as the potential for Cl2 evolution is considerably more anodic than evolution of CO or CO2, as shown in Fig. 3. Moreover, it is more difficult to electro-deoxidize the oxides and it is more favourable to form Al2Ca at 700 °C than 900 °C.
3.2 Electrolytic reduction of solid Al2O3
In this work, the progress of reaction is described by the extent of reduction (X) defined as follows:
where X is the extent of reduction, W 0 is the mass of oxide (Al2O3) at the beginning (t = 0), and W t is the mass of oxide or oxides at time t. A series of electrolysis experiments were undertaken under various conditions to investigate key factors influencing the direct electrolytic reduction of solid Al2O3. The experimental conditions used and the values of X obtained are given in Table 1.
Figure 4 shows the typical current-time curve measured at the early stage of electrolysis of the fully dense tube of Al2O3* in the fused CaCl2–NaCl electrolyte containing 32.2 mol% NaCl at 900 °C (run 1). It was found from Fig. 4 that the currents, on the application of 3.1 V, jumped to a value of 0.9 A and then decayed rapidly, followed by steady current decline with time, approaching the residual values of around 0.2 A after 15 h electrolysis. This is similar to the current-time behaviours reported for direct electrochemical reduction of SiO2 and TiO2 in molten CaCl2 at 850 °C [16, 23]. Cowley reported that solubilities of Ca–Na alloys in approximately equimolar CaCl2–NaCl melts are 0.4–4.5 wt% Na, increasing with increasing temperature, and the Ca–Na alloys contain at least 98 at% Na at 500–700 °C [44]. Dissolution of elemental Ca and Na into the CaCl2–NaCl melt imparted certain degrees of electronic conductance to the melt [45]. The observed residual currents were due to small amounts of dissolved Ca and Na being generated during electrolysis. Moreover, solid Al2O3 is regarded as an electrical insulator since its electrical resistivity is 2 × 104 Ω m at 1,000 °C [46]. Consequently, the cathodic ionization of the oxygen from the Al2O3 exposed to the electrolyte was restricted to the Al2O3/electrolyte/Nb three phase boundaries (3PBs) at 3.1 V.

Current–time curve of the Al2O3 tube during electro-deoxidation at 3.1 V in the molten CaCl2–NaCl electrolyte containing 32.2 mol% NaCl at 900 °C
Figure 5 shows the photographs of (a) the Al2O3 tubes before and after electrolysis (run 1), (b) the metal droplets collected from the Nb basket after 66 h electrolysis, and (c) the inner surface of the Nb sheet employed for fabricating the basket after electrolysis, respectively. As observed from Fig. 5a, the size of the Al2O3 tube diminished in three dimensions after electrolysis, implying that electronically conducting phases had formed on the Al2O3 surface which subsequently acted as electron transfer medium for the electro-deoxidation to occur not only at the 3PBs of the oxide/electrolyte/Nb but also at the oxide/electrolyte interface. As a result, the reduction spread along the surface of the tube and then into the interior. Ionized O2− dissolved into the electrolyte and liquid Al formed which spheroidized and the droplets fell from the tube to the basket bottom. It is also observed from Fig. 5b that the nearly spherical droplets collected had droplet sizes ranging from several hundred microns to about 2 mm, indicative of coalescence amongst the initial small-sized droplets during electrolysis. EDS spectra of the droplet surfaces showed predominant peaks for Al and a very weak peak for O, indicating that the surface material was Al. Figure 5c shows that some droplets had reacted with the Nb basket, forming new phases consisting of Al and Nb (marked by A) on the inner surface of the Nb basket (marked by B).

Photographs of a the dense tubes of Al2O3 before and after 66 h electrolysis in the CaCl2–NaCl melt consisting of 32.2 mol% NaCl at 900 °C (run 1), b the reduced Al droplets collected from the Nb basket after electrolysis, and c the Nb sheet used for fabricating the Nb basket after electrolysis
The above results confirmed that the electrolytic reduction of the oxides to liquid Al was responsible for the current–time behaviour in Fig. 4. The value of X, as determined from the measured mass change of the Al2O3 before and after electrolysis, was calculated to be 24% according to Eq. 5. In this work, the cathodic current efficiency was defined by a percentage of the measured mass change in O content of an oxide cathode before and after electrolysis compared to the total O mass change evaluated from the measured current–time curve. The cathodic current efficiency over the period of 66 h electrolysis was determined to be about 25% after the residual current subtraction.
Figure 6a illustrates the SEM backscattered electron (BE) micrograph of the polished cross section of one of the droplets in Fig. 5b. The droplet is characterized by distinct gray and bright areas, typical of microstructure of the eutectic-type alloys, and is fully covered with a grey layer. Figure 6b is the enlarged SEM-BE image of that in Fig. 6a, clearly showing that the large grey areas marked by A is surrounded by mixed grey and bright areas marked by B. An EDX analysis of the layer marked C and the grey areas gave 98 at% Al and 2 at% O, while the mixed grey and bright areas had 81 at% Al, 17 at% Ca, and 2 at% O. As evaluated from the Al–Ca phase diagram [47], Al should co-exist with Al4Ca (20 at% Ca) at temperatures below 613 °C. Therefore, the mixed grey and bright areas correspond to a mixture of the Al and Al4Ca, with the detected O stemming from surface oxidation of the sample at room temperature. Elemental Na was not detected by EDS and EDX analyses of all the areas within the sample in Fig. 6a.

The overall composition of the remaining droplets in Fig. 5b, as determined by ICP, was 98.2 at% Al and 1.8 at% Ca. The ICP result confirmed that the oxides had been reduced to Al-rich Al–Ca alloys at 900 °C. With the overall Ca content (1.8 at% Ca) being far less than that in Al2Ca (33 at% Ca), it is believed that the reduction of the oxides proceeds in the electrolyte where activities of the dissolved Ca are less than 2.43 × 10−8, as predicted from Fig. 2b; otherwise, the Ca dissolved in the electrolyte in equilibrium with solid Al2Ca would diffuse into the reduced Al driven by differences in Ca chemical potential between the electrolyte and Al to produce Al2Ca. Reaction 1 is hence predominant at the cathode. During cooling of the cathode products after electrolysis, the solid Al4Ca and Al are precipitated from the Al-rich Al–Ca melt, leading to the observed eutectic-type alloy microstructure in Fig. 6.
3.3 Intermediate reaction products
Figure 7a shows the SEM-BE micrograph of the polished cross section of the small Al2O3 tube electrolysed in the equimolar CaCl2–LiCl melt at 900 °C (run 2), from which it can be clearly observed that the un-reduced core was fully covered with a dense layer. An EDS analysis of several spots within the core and the layer revealed that the core remained as Al2O3, while the layer was composed of elemental Al, Ca, O, and Cl. Figure 7b shows the enlarged SEM-BE image taken from a portion of the layer in Fig. 7a. As observed from Fig. 7b, the layer actually consists of grey areas, bright grey areas, and bright areas, as marked by A, B, and C, respectively. The EDX data measured from the different spots of the individual areas are given in Table 2. Elemental Li has an atomic weight too small to be detected by EDS and EDX. These results indicate the formation of aluminates within the layer. EDS analysis was also performed on the cross section of the layer on the Al2O3 after electrolysis in the CaCl2–NaCl melt (run 3). The obtained EDS spectra, in this case, exhibited peaks for elemental Al, Ca, O, and Cl, but no peaks for elemental Na within the layer.

SEM backscattered electron (BE) images of a the polished cross section of the remaining part of the Al2O3 tube after 23.7 h electrolysis in the equimolar melt of CaCl2–LiCl at 900 °C (run 2) and b enlarged SEM-BE image showing a portion of the layer formed on the core of the un-reduced Al2O3 tube in Fig. 7 a
Figure 8 shows the XRD spectrum of the isolated layer on the Al2O3 tube in Fig. 7, which reveals that the layer consists of metallic Al, Ca12Al14O33, and Ca12Al14O32Cl2 as the major phases, LiAlO2 as the medium phase, and CaO or CaO2 as the trace or minor phase, free of Al2O3. These are to be expected as the standard Gibbs free energy change for the reaction of a Na aluminate with CaCl2 to form a Ca aluminate and NaCl is negative at 900 °C. For example, Na2Al2O4 + CaCl2 = CaAl2O4 + 2NaCl, \( \Updelta {\text{G}}_{{1173{\text{K}}}}^{0} = - 99.271\;{\text{kJ}}\;{\text{mol}}^{ - 1} \) should take place at 900 °C, with the equilibrium constant \( k_{p} = 2.633 \times 10^{4} \)[41]. In contrast, the standard Gibbs free energy change for the reaction of LiAlO2 with CaCl2 to form CaAl2O4 and LiCl is only slightly negative (\( \Updelta G_{1173K}^{ \circ } = - 4.087\,{\text{kJ}}\,{\text{mol}}^{ - 1} \)), with a value of k p = 1.52 at 900 °C [41]. The XRD result confirms that the aluminates were formed in situ on the Al2O3 surface and were then electro-deoxidized to Al-rich Al–Ca alloys mostly at the aluminate/electrolyte interfaces. Ca12Al14O32Cl2 and partially reduced Ca aluminates are electronic conductors [48–50]. For example, a partially reduced single crystal of Ca12Al14O33 has an electronic conductivity of about 1 S m−1 at 27 °C [48]. The presence of very fine Al droplets maximises the 3 PB contact between the aluminate/electrolyte/Al within the layer, thus promoting electro-deoxidation. An advancing Al2O3/electrolyte reaction front to form the aluminates moves inwards and the un-reacted Al2O3 core shrinks continuously as the electro-deoxidation progresses, with the fine liquid Al coalescing, resulting in large droplets falling from the tube surface.

XRD spectrum of the layer formed on the Al2O3 tube after electrolysis in the equimolar CaCl2–LiCl electrolyte at 900 °C (run 2)
According to Figs. 2 and 3, the magnitude of pO2− should determine which aluminate is stable within the layer on the Al2O3 surface in contact with electrolyte. The CaCl2 · 2H2O and anhydrous LiCl after thermal drying in air contained about 0.595 mol% CaO and 1.77 × 10−3 mol% Li2O, respectively, as measured using the standard acid-base titration method. Mole fractions of CaO are then calculated to be 4.03 × 10−3 in the CaCl2–NaCl melt containing 32.2 mol% NaCl and approximately 2.98 × 10−3 in the equimolar CaCl2–LiCl melt when neglecting the initial Li2O amount in the LiCl. The Henrian activity coefficients of CaO (\( {{\upgamma}}_{{\left( {\text{CaO}} \right)}} \)) as a function of temperature were recently determined experimentally by Wang et al. for molten CaCl2 and equimolar CaCl2–NaCl and CaCl2–LiCl melts saturated with CaO [43]. The temperature dependence of \( {{\upgamma}}_{{\left( {\text{CaO}} \right)}} \) obtained by Wang et al. may be applicable to dilute solutions of CaO in the molten salts by assuming that \( {{\upgamma}}_{{\left( {\text{CaO}} \right)}} \) is independent of CaO concentration (Henry’s law) and changes in NaCl concentration from 50 mol% to 32.2 mol%. The extrapolated values of \( {{\upgamma}}_{{\left( {\text{CaO}} \right)}} \) in the CaCl2–NaCl melt were calculated according to their equations [43] to be 13.71 at 700 °C and 12.59 at 900 °C, while those in the equimolar CaCl2–LiCl melt were 1.41 at 700 °C and 1.36 at 900 °C. Thus, the values of pO2− before pre-electrolysis were estimated at 1.26 at 700 °C and 1.29 at 900 °C in the CaCl2–NaCl melt and were 2.38 at 700 °C and 2.29 at 900 °C for the equimolar CaCl2–LiCl melt. The calculations show that it is thermodynamically possible for CaO in the present electrolytes without pre-electrolysis to react with Al2O3 to produce Ca12Al14O33 or Ca12Al14O33 and LiAlO2 over these pO2− regions, as predicted from Figs. 2 and 3. However, the CaO or Li2O dissolved in the electrolytes are decomposed to evolve CO/C2 gases at the carbon anode and to deposit cathodically metallic Ca or Li at the surface of the stainless steel crucible exposed to the electrolytes during pre-electrolysis at 2.8 V. The reduced Ca or Li subsequently dissolves in the CaCl2-based electrolytes. The amount of the Ca or Li reduced from its oxide initially present in the salt is very low because of the very low quantities of initial oxide in the thermally dried salt. The electrochemical removal of the O2− dissolved initially in the electrolytes by pre-electrolysis significantly reduced the O2− activities, thus increasing values of pO2− in the electrolytes.
From the thermodynamic predictions and observation of the various phases present in the products it can be concluded that the following reactions take place:
Li aluminate may also be formed by the reaction
and can be decomposed by the reaction:
The potentials for these reactions can be compared with the potential for the reduction of CaO:
3.4 Factors affecting reduction behaviour
3.4.1 Effect of Al2O3 surface area
The specific surface area of the small Al2O3 tubes employed in run 3 was calculated to be 5.2 × 10−4 m2 g−1, greater than that of the large Al2O3* tube (3.5 × 10−4 m2 g−1) as used in run 1. As given in Table 1, the magnitudes of X achieved were 24% for run 1 and 71% for run 3. It was found that a 33% rise in specific surface area of the Al2O3 tubes led to an increase in the X value by 66%, although the electrolysis duration of run 3 was almost three times shorter than that of run 1. The findings suggested that the electro-deoxidation could be effectively enhanced by enlarging specific surface area of solid Al2O3.
3.4.2 Effect of electrolyte system
Table 1 shows that the value of X for run 2 using the equimolar CaCl2–LiCl electrolyte was 78% and was only slightly higher than X = 71% from run 3 with the CaCl2–NaCl electrolyte. The results indicate that the use of the CaCl2–LiCl electrolyte has no obvious enhancement effect on the overall rate of electrolytic reduction of Al2O3 compared to that of CaCl2–NaCl electrolyte at 900 °C under these conditions. The metallic droplets collected after run 2 had similar sizes to those from run 1 or 3 and they were actually Al-rich Al–Ca alloys containing 5.4 at% Ca on average, as determined by ICP. This amount of Ca was higher than that in the Al produced from run 1, although the initial CaO content (0.298 mol% CaO) in the equimolar CaCl2–LiCl melt was lower than that (0.403 mol% CaO) in the CaCl2–NaCl melt used in this work. This is probably due to two factors. First, the dissolved O2− is predominantly associated with the Ca2+ as (CaO) instead of Na+ as (Na2O) or Li− as (Li2O) in the electrolytes:
where (CaO), (Na2O), and (Li2O) are the CaO, Na2O, and Li2O dissolved in the electrolyte melts, respectively. The equilibrium constants (k p ) for Reactions 11 and 12 at 900 °C are 1.196 × 109 and 2.133 × 102 [41], respectively, indicating that the activities of Li2O and especially Na2O should be much lower than that of CaO in the electrolytes. The CaO dissolved in the electrolytes then reduces to metallic Ca at reduced activity through Reaction 10 during electrolysis and the reduced Ca subsequently dissolves in the CaCl2-based electrolyte since Ca is soluble in molten CaCl2 (2.1–3.6 mol% Ca in molten CaCl2 at 800–940 °C [51]). Second, equilibria between (Ca) and (Na) in the CaCl2–NaCl melt and between (Ca) and (Li) in the equimolar CaCl2–LiCl melt may be governed by the following exchange reactions:
where (Ca), (Na), and (Li) are the Ca, Na, and Li dissolved in molten CaCl2–NaCl and CaCl2–LiCl mixtures, respectively. Solubilities of Na in molten NaCl are 2.8–10.2 mol% Na at 809–890 °C [52], while those of Li in molten LiCl range from 0.66 mol% to 1.22 mol% Li at 662–850 °C [53]. The equilibrium constants (k p ) for Reaction 13 are 44.66 at 700 °C and 59.17 at 900 °C, whereas the values of k p for Reaction 14 are only 1.91 × 10−2 at 700 °C and 1.335 × 10−2 at 900 °C [41]. These indicated that activities of the Ca dissolved in the equimolar CaCl2–LiCl melt are much higher than those in the CaCl2–NaCl melt used. The higher activities of the Ca dissolved in the CaCl2–LiCl melt cause the observed higher Ca content in the reduced Al. Nevertheless, the Ca activity in the CaCl2–LiCl melt is still below \( {\text{a}}_{\text{Ca}} \) [Al2Ca(s)] = 2.43 × 10−8 under the present conditions according to Fig. 3b. Although Na activities in the CaCl2–NaCl melt might be greater than those of Ca, elemental Na was undetectable by EDS or EDX in the Al reduced using this electrolyte, probably because of the very low Na solubilities in liquid Al (e.g., about 0.18 at% Na in molten Al at 665 °C [54]).
The different values of X were obtained from runs 4 and 7 conducted at 700 °C using the CaCl2–NaCl and molten LiCl electrolytes. It was found that the Al2O3 tube was reduced at 700 °C, with X = 35% obtained after 23.33 h electrolysis in the CaCl2–NaCl melt (run 4) as opposed to X = 71% at 900 °C in run 3. It was, however, observed that no electro-deoxidation of the Al2O3 took place in the molten LiCl at 700 °C (run 7). Two main causes were considered to be responsible for the latter observation. First, at lower temperatures, probably 700 °C or less, there are increasing effects of mass transport constraints on the O to be ionized from insulating solid Al2O3, hence the very sluggish kinetics of oxygen transport within the Al2O3 lattice and consequently solid-state cathodic oxygen ionizations from the Al2O3 at the 3PBs of Al2O3/LiCl/Nb. The diffusivity of oxygen in α-Al2O3 is very low and it decreases drastically with decreasing temperature, e.g., from about 2.2 × 10−22 m2 s−1 at 1,400 °C down to 1.1 × 10−24 m2 s−1 at 1,200 °C [55]. As to the formation of LiAlO2 on the Al2O3 surface, unfortunately, there are so far no activity coefficients (\( {{\upgamma}}_{{\left( {{\text{Li}}_{ 2} {\text{O}}} \right)}} \)) of Li2O dissolved in molten LiCl available in the literature. It is thus difficult to estimate initial O2− activity (i.e., pO2−) in the LiCl melt and predict the LiAlO2 formation at 700 °C. Also, any change in the Al2O3 mass before and after electrolysis was not measurable. Second, even if a thin layer of LiAlO2 forms on the Al2O3 surface, LiAlO2 is much more stable than the Ca aluminates as evident from Fig. 3a. As discussed previously, LiAlO2 is even thermodynamically more stable than molten LiCl over the pO2− region of 0–2.2 at 700 °C. It is thus difficult for cathodic ionization of the oxygen from the LiAlO2 to occur in the LiCl melt at 700 °C.
Overall, the present results reveal that the CaCl2–NaCl electrolyte is more suitable than the equimolar CaCl2–LiCl electrolyte in terms of Al purity. Although molten CaCl2 or LiCl has appreciable solubility for O2−, the use of CaCl2 as the main electrolyte component appears to facilitate the electro-deoxidation of Al2O3 more effectively than LiCl.
3.4.3 Effect of electrolysis temperature
It is well known that fused salt electrolysis at low temperatures may offer a number of advantages, e.g., lower corrosive damage to the cell vessel, lower loss of electrolyte due to vaporization, and wide availability of materials for electrodes and cell linings. Attempts were therefore made to conduct the electrolytic reduction of solid Al2O3 at 700 °C (runs 4–9).
For the CaCl2–NaCl melt containing 32.2 mol% NaCl, the value of X for run 4 conducted at 700 °C was 35% and was about half of that achieved for run 3 at 900 °C. The observed increase in X with increasing temperature can mainly be ascribed to decreasing stability of the oxides in the electrolyte, as evident from Fig. 2, and enhanced kinetics of Reactions 6 and 7.
However, when using the equimolar CaCl2–LiCl electrolyte, the value of X = 100% was obtained from run 5 after electrolysis at 700 °C, whereas the value of X was 78% for run 2 using the same electrolyte at 900 °C. Complete reduction was repeated by conducting run 6 under almost the same conditions as run 5. For both runs 5 and 6, the cathode product was Al2Ca only, free of any oxides, whereas the products from run 2 at 900 °C were metallic Al with 5.4 at% dissolved Ca and the un-reduced oxides. Liu and Poignet reported that Li activities increase with decreasing temperature at temperatures between 650 and 800 °C for dilute solutions of Li in Li–LiCl melts [56]. Therefore, the activities of Li and Ca dissolved in the CaCl2–LiCl melt become larger as the melt temperature decreases because of increasing Li activity and consequently Ca activity through Reaction 14 with decreasing temperature. A comparison in iso-activity lines of Ca for the Al/Al2Ca equilibrium between 700 and 900 °C in Fig. 3 reveals that the activity of the Ca dissolved in the electrolytes needed to form Al2Ca at 700 °C is much lower than that at 900 °C, suggestive of the easier formation of Al2Ca at 700 °C than 900 °C. This prediction is consistent with the findings from runs 2 and 5 or 6. The findings also indicate that the activities of Ca dissolved in the equimolar CaCl2–LiCl melt had exceeded the value for Al2Ca to be thermodynamically stable at 700 °C and were below the value for Al2Ca to be stable at 900 °C, which explains the formation of Al2Ca in the cathode product at 700 °C but not at 900 °C. The observed faster reduction rates at 700 °C compared to 900 °C probably result from a small degree of contributions from chemical reduction of the Al2O3 by the dissolved Ca and Li.
To clarify possible involvement of metallothermic reduction in the overall reduction process, especially at low temperatures, electrolysis was carried out in a Li-saturated Li–LiCl electrolyte at 700 °C (runs 8 and 9). It was found that the reduction of the Al2O3 occurred, with the reduction extent increased and the reduction rates decreased with time. It should be mentioned that, in a Li-saturated LiCl–Li melt, the electronic conductivity of the melt is very high and very little ionic transfer occurs in the melt. Any contributions from electro-deoxidation of the Al2O3 to the overall reduction might be limited. As a result, the observed reduction can be mainly attributed to the reaction:
The experimental results indicate that the chemical reduction of the Al2O3 by the Ca and Li dissolved in CaCl2–LiCl melt might be involved in the overall reduction process provided that the activities of Ca and Li dissolved in the melt are increased significantly.
This study shows that the in situ formation of the electronically conducting oxides on the Al2O3 surface, determined by O2− activities in the electrolytes, is essential for the electro-deoxidation of solid Al2O3 to continue. The O2− activities and thermodynamic stabilities of the oxides are closely related to the salt systems used and appear to be important in determining observed reduction behaviour and Ca contents of the reduced Al. The Al metal with small amounts of Ca produced can be directly used as the deoxidant for the production of steel and Ti aluminides or as the master alloys for the production of various advanced alloys containing Al and Ca constituents [57, 58]. This may be economically attractive because of the higher prices of Ca than Al. Alternatively, high-purity Al can be produced from the present products using existing melt purification techniques, such as chlorination, which is being utilized for the efficient removal of Na from Al metal produced by the Hall–Heroult process.
4 Conclusions
Solid Al2O3 was reduced electrolytically to Al metal in molten CaCl2–NaCl and CaCl2–LiCl electrolytes, respectively, at an applied potential of 3.1 V and at 900 °C. It was found that the overall rates of direct electrolytic reduction of the solid Al2O3 were little influenced by the electrolyte systems used at 900 °C. It was also found that lower concentration of Ca in the reduced Al was obtained with molten CaCl2–NaCl electrolyte compared to that with CaCl2–LiCl electrolyte. The Ca aluminates were formed as the main intermediate reaction products and were subsequently reduced electrolytically to form Al-rich Al–Ca alloys during the course of electrolysis, which was partly responsible for the presence of Ca in the Al produced. The formation of Al–Ca alloys was likely to be caused by Ca being produced at reduced activity at the cathode surface and dissolving in the molten Al. The experimental observations were consistent with the thermodynamic predications using E-pO2− diagrams of Al in the electrolytes at 700 and 900 °C.
References
Grjotheim K, Welch BJ (1988) Aluminium smelter technology. Aluminium-Verlag, Dusseldorf
Kirk-Othmer (2002) Encyclopaedia of chemical technology, vol 2. Wiley, New York, p 279
Fray DJ (1988) In: Sohn HY, Geskin ES (eds) Metallurgical processes for the year 2000 and beyond. TMS, Warrendale, PA, p 493
Edwards L, Kvande H (2001) J Met 53:28
Welch BJ, Hyland MM, James BJ (2001) J Met 53:13
Tabereaux AT (1992) J Met 44:20
Choate WT, Green JAS (2003) U.S. energy requirements for aluminum production: historical perspective, theoretical limits and new opportunities. U.S. Department of Energy, Energy Efficiency and Renewable Energy, Washington, DC
Cochran CN (1987) Production of aluminium by alternate processes. In: 8th international light metals congress, Leoben, Vienna, p 82
Fray DJ, Farthing TW, Chen Z (1998) International patent PCT/GB99/01781, first filing date, 05 June
Chen GZ, Fray DJ, Farthing TW (2000) Nature 407:361
Fray DJ (2000) Metall Mater Trans B 31B:1153
Fray DJ (2001) J Met 53:26
Fray DJ (2002) Can Metall Q 41:433
Yan XY, Fray DJ (2002) Metall Mater Trans B 33B:685
Nohira T, Yasuda K, Ito Y (2003) Nat Mater 2:397
Jin XB, Gao P, Wang DH, Hu XH, Chen GZ (2004) Angew Chem 116:751
Yan XY, Fray DJ (2005) J Electrochem Soc 152:D12
Yan XY, Fray DJ (2005) J Electrochem Soc 152:E308
Qiu GH, Ma M, Wang DH, Jin XB, Hu XH, Chen GZ (2005) J Electrochem Soc 152:E328
Schwandt C, Fray DJ (2005) Electrochim Acta 51:66
Sakamura Y, Kurata M, Inoue T (2006) J Electrochem Soc 153:D31
Yan XY, Fray DJ (2005) Adv Funct Mater 15:1757
Jiang K, Hu XH, Ma M, Wang DH, Qiu GH, Jin XB, Chen GZ (2006) Angew Chem 118:442
Wenz DA, Johnson I, Wolson RD (1969) J Chem Eng Data 14:252
Story JB, Clarke JT (1957) J Met 9:1449
Littlewood R (1962) J Electrochem Soc 109:525
Ferry DM, Picard GS, Tremillon BL (1988) Trans IMM C 97:C21
Martinez AM, Castrillejo Y, Barrado E, Harrberg GM, Picard G (1998) J Electroanal Chem 449:67
Dring K, Dashwood R, Inman D (2005) J Electrochem Soc 152:D184
Dring K, Bhagat R, Jackson M, Dashwood R, Inman D (2006) J Alloys Comp 419:103
Bhagat R, Jackson M, Inman D, Dashwood R (2008) J Electrochem Soc 155:E63
Yasuda K, Nohira T, Hagiwara R, Ogata YH (2007) J Electrochem Soc 154:E95
Picard G, Seon F, Tremillon B (1980) Electrochim Acta 25:1453
Picard G, Seon F, Tremillon B (1983) Selective chlorination of oxides in suspension in molten chlorides. In: Proceedings of 1st inter symposium on molten salt chemistry and technology, Kyoto, p 49
Tremillon B, Picard G (1987) In: Mamantov G, Mamantov R (eds) Molten salt chemistry. D Reidel Publishing Company, Boston, MA, p 305
Boghosian S, Godo A, Mediaas H, Ravlo W, Ostvold T (1991) Acta Chem Scand 45:145
Stern KH, Panayappan R, Flinn DR (1977) J Electrochem Soc 124:641
Janz GJ (1967) Molten salts handbook. Academic Press, New York, p 180
Roth RS (ed) (2001) Phase equilibria diagrams, vol XIII. The American Ceramic Society, Ohio, p 84
Chartrand P, Pelton AD (2001) Metall Mater Trans A 32A:1361
HSC (1999) Outokumpu chemistry for windows, version 4.0, Outokumpu Research Oy Information Service, Pori, Finland
Johnson GK, Pierce RD, Poa DS, McPheeters CC (1994) In: Mishra B, Averill WA (eds) Actinide processing: methods and materials. TMS, Warrendale, PA, p 199
Wang SL, Zhang FH, Liu XA, Zhang LJ (2008) Thermochim Acta 470:105
Cowley WE (1982) In: Lovering DG (ed) Molten salt technologies. Plenum Press, New York, p 57
Haarberg GM, Thonstad J (1989) J Appl Electrochem 19:789
Shackelford JF, Alexander W (2001) CRC Materials science and engineering handbook, 3rd edn. CRC Press, FL, p 565
Okamoto H (2003) J Phase Equilib 24:91
Matsuishi S, Toda Y, Miyakawa M, Hayashi K, Kamiya T, Hirano M, Tanaka I, Hosono H (2003) Science 301:626
Kim SW, Matsuishi S, Nomura T, Kubota Y, Takata M, Hayashi K, Kamiya T, Hirano M, Hosono H (2007) Nano Lett 7:1138
Medvedeva JE, Teasley EN, Hoffman MD (2007) Phys Rev B 76:155107
Sharma RA (1970) J Phys Chem 74:3896
Bredig MA, Johnson JW, Smith WT (1955) J Am Chem Soc 77:307
Nakajima T, Minami R, Nakanishi K, Watanabe N (1974) Bull Chem Soc Jpn 47:2071
Murray JL (1983) Bull Alloy Phase Diagr 4:137
Heuer AH, Lagerlof KPD (1999) Phil Mag Lett 79:619
Liu J, Poignet JC (1990) J Appl Electrochem 20:864
Kirk-Othmer (1992) Encyclopedia of chemical technology, 4th edn. Wiley, New York, p 783
Ozturk K, Zhong Y, Luo AA, Liu ZK (2003) J Met 55:40
Acknowledgments
The authors gratefully acknowledge the Light Metals Flagship, a National Research Program of Australia, for financial support. Mrs. N. A. Olshina is thanked for conducting XRD analyses of the samples. Characterization of the samples using the SEM-EDS and EDX by Mr. D. J. Cameron and Dr. A. M. Glenn is appreciated. Assistance from the Analytical Services Group of CSIRO Minerals at Clayton is also acknowledged. Finally, the authors would like to thank the reviewer for the valuable comments that were considered in revising and improving our manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yan, X.Y., Fray, D.J. Direct electrolytic reduction of solid alumina using molten calcium chloride-alkali chloride electrolytes. J Appl Electrochem 39, 1349–1360 (2009). https://doi.org/10.1007/s10800-009-9808-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-009-9808-3