
Abstract
Using cyclic-voltammetric techniques, a pyrophosphate-cyanide electrolyte for the electrodeposition of compact Ag–Sn alloy coatings is investigated. This electrolyte is suited to further investigations on the alloy composition, structure and properties. The electrodeposition of coatings with up to 40 wt% Sn is possible from the investigated complex electrolytes. The alloy surpasses the saturation limit of the silver lattice with Sn and allows the formation of coatings with phase heterogeneity. At high tin content an ordered spatial distribution of different alloy phases on the cathodic surface can be observed. The pattern formation in this system looks very similar to the phenomena and structures observed during electrodeposition of other silver alloys, such as Ag–Sb, Ag–Bi and Ag–In.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Electrodeposited silver finds wide application in contemporary technology due to its excellent properties such as high electrical and thermal conductivity, high reflexion coefficient, and excellent catalytic and antibacterial properties [1]. Alloying with other metals can achieve an enhancement of some properties of silver, like low hardness, wear and tarnish resistance. Several electrolytes and processes for electrodeposition of silver alloys with antimony [2–10], copper [5, 11–14], cadmium [5, 12, 15–19], bismuth [3, 20–24], lead [5, 12, 25–27], indium [3, 5, 28–35], tin [36–39] and other metals have been reported.
In most cases, electrodeposits which contain the alloying element up to several weight percents are of practical importance. In such alloys an enhancement in the physico-mechanical properties of the basic metal is observed, especially in the region of solid solution formation [40]. Coatings with increased content of the alloying element could be of increased scientific interest when simultaneous different phases of the alloy system are formed during deposition. In such cases self-organization phenomena and formation of ordered spatio-temporal structures on the cathode surface may be observed. Such structure and pattern formation occurs during co-deposition of silver with antimony [41–43], indium [28, 29, 32], and bismuth [22].
Silver alloys with tin are important because of their improved corrosion and tarnish resistance (at high silver contents in the alloy) compared to pure silver, as well as for their better physico-mechanical properties and increased melting point (at high tin contents) compared to pure tin, the latter being important when several contact elements for electronics are plated. The physical properties of Ag–Sn alloys were established a long time ago [44]. The best and relatively long-lasting strength properties are observed in the eutectic alloy [45]. Alloys with 2.5–5 wt% Ag show higher resistance to plastic deformations compared to pure tin [45], and alloys with 11 wt% Ag form solder joints with 2–4 times higher strength than pure tin or tin-lead alloys [46]. Alloys with 5–7 wt% Sn have a significantly better tarnish resistance compared to pure silver [47].
The difference between the standard potentials of Ag and Sn is large (0.94 V) and therefore the alloy can be deposited only from electrolytes where silver is bonded in strong complexes. As possible complex forming agents CN−, SCN−, S2O3 2− and S2O8 2− anions are mainly used. The reduction potentials of silver from these complexes are within the interval 0.21 to −0.3 V versus the normal hydrogen electrode. Electrodeposition of the alloy is possible from electrolytes on a different basis. Literature data exist about the application of alkaline [48], cyanide [49] cyanide-pyrophosphate [36, 39, 50–52], iodide-pyrophosphate [53, 54] and acid electrolytes, the latter based on salts of both metals and thiourea as a complex forming agent for silver [55], and methanesulphonic [56], mercaptopropionsulphonic [57] and other electrolytes. There are variants for both tin-rich, as well as tin-poor coatings and it is possible, under certain conditions, to achieve simultaneous deposition of phases with different tin content, which can self-organize to form ordered periodic structures on the cathode surface.
The aim of the present work is to find, on the basis of cyclic-voltammetry, an electrolyte for the electrodeposition of compact Ag–In alloy coatings, appropriate for further investigations on their composition, structure and properties, as well as to find conditions for the formation of periodic spatio-temporal structures on the electrode surface.
2 Experimental
The composition of the investigated pyrophosphate-cyanide electrolytes is given in Table 1. All solutions were made with p. a. purity chemicals and distilled water.
Both salts SnCl2 and K4P2O7 were dissolved separately in water and the tin containing solution was added to the pyrophosphate one under continuous stirring with a magnetic stirrer. Then KOH was added to the solution in small portions and the electrolyte became milky white. Na2EDTA · 2H2O was then added in small portions, so that the electrolyte became clear. The next step involved the addition of H2O2 and, after half an hour, the last solution containing KAg(CN)2 and KCN was added. In some electrolytes Sn2P2O7 was used instead of SnCl2 · 2H2O.
The experiments were performed in a 50 cm3 three-electrode glass cell at room temperature without agitation of the electrolyte. The vertical working electrode (area 0.6 cm2) and the two counter electrodes were made of platinum. An Ag|AgCl reference electrode (EAg|AgCl = −0.197 V vs. SHE) was used. The reference electrode was placed in a separate cell filled with 3 M KCl solution (Merck), connected to the electrolyte cell by a Haber-Luggin capillary through an electrolyte bridge also containing 3 M KCl solution. All potentials are given against this reference electrode.
The experiments were carried out at room temperature by means of a computerized PAR 263 A potentiostat/galvanostat using the Soft Corr II software. The sweep rate was varied between 0.005 and 1 V s−1.
The Sn percentage in the coatings was determined by X-ray fluorescence analysis (Fischerscope X-RAY HDVM-W). The surface morphology of the coatings was studied by optical and scanning electron microscopy (SEM). The phase analysis was performed by means of a Philips PW 1050 diffractometer at 2θ angles from 25 to 80° with CuKα irradiation and a scintilation detector.
3 Results and discussion
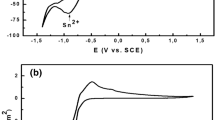
The hydrogen evolution in the absence of Ag and Sn in the basic electrolyte starts at about −1,000 mV (Fig. 1, curve 1). Silver deposition starts in the absence of Sn at similar potentials (curve 2). Two cathodic maxima are registered at about −1,030 and −1,174 mV possibly due to the deposition of silver from different complexes [58] and the silver dissolution reaction runs in one stage starting at about −730 mV reaching its maximum rate at a potential of −450 mV.

Cyclic voltammetric curves: 1 (-.-.-.), BE without Ag and Sn; 2 (….), BE without Sn; 3 (- - - -), BE without Ag; 4 (——), BE; sweep rate 25 mV s−1
One cathodic maximum at about −1,190 mV is observed during electrodeposition of tin (curve 3) in the absence of silver, i.e. in the region of the second cathodic maximum of silver, which offers the opportunity for co-deposition of both metals.
Two tin dissolution maxima are observed in the anodic cycle at about −840 and −640 mV. The first one is much more negative than the silver dissolution potential in this electrolyte. The second cathodic maximum is in the region of silver dissolution. In this system, silver is a more positive element than tin. The electrodeposition of the alloy is of regular type according to Brenner [12].
In the presence of both metals in the electrolyte (curve 4), silver is deposited at more negative potentials (−1,060 mV) compared to deposition in the tin-free electrolyte (−1,030 mV). The first cathodic maximum in the cyclic voltammetric curve corresponds to pure silver. At the position of the second cathodic maximum of silver (see curve 2), as well of the cathodic maximum of tin (see curve 3) a hump in the curve appears. At this potential a silver–tin alloy coating is probably deposited. Tin is deposited in the presence of silver with a polarization of about 130 mV and its deposition potential shifts −1,190 to −1,320 mV (see curve 3).
In the anodic part of the cyclic voltammetric curve three electrochemical reactions are registered. The first two of them, at about −580 and −430 mV, respectively, appear in the silver dissolution region, i.e. the alloy coating is dissolved at more positive potentials than pure tin. There is no maximum that can be ascribed to the tin phase. i.e. tin is probably combined in alloy phases with silver and the alloy coating is nearly as noble as the silver itself. Another possibility is that pure tin in the coating is covered by silver due to a replacement reaction when the system passes over the potential range of the tin dissolution reaction in the sweep in the anodic direction. The third anodic maximum at about +400 mV probably corresponds to the dissolution of passive films formed at more negative potentials in the anodic cycle.
In order to ascribe the anodic peaks to the different phases of the alloy system electrodeposited in the cathodic cycle, the potential scan direction was changed at different cathodic potentials within the interval −1,060 to −1,400 mV.
When the potential sweep direction is changed in the region of the first cathodic maximum, i.e. in the region of pure silver deposition, the anodic maximum corresponds to the dissolution of this metal (Fig. 2). When the scan direction is changed in the region of the hump in the curve after the first cathodic maximum, i.e. in the region where the co-deposition of both metals and the alloy formation is possible, the electrodeposit is dissolved at slightly more positive potentials and the silver dissolution peak becomes a hump on the left side of the maximum. The X-ray diffraction spectrum (Fig. 3, solid line) of a sample deposited at a potential of −1,235 mV (see Fig. 2, curve 3) shows only the peaks of the silver lattice with some shift in low 2θ angle direction that corresponds to the expansion of the silver lattice by tin atoms when the solid solution of tin in silver (α-phase) is formed. The absence of a tin dissolution peak in the CV curves (Fig. 2, curve 3) confirms that all the co-deposited tin is included in the α-phase.

Cyclic voltammetric curves in BE Vertex potentials: −1,060 mV (——); −1,136 mV (− − −); −1,235 mV (....); −1,263 mV (-. -.); −1,300 mV (-.. -..); −1,400 mV (—-); sweep rate 25 mV s−1

XRD of Ag–Sn coatings deposited in BE at constant potential for 1 min: (——) E = −1,235 mV; (- - -) E = −1,400 mV; (...) after a repetitive scanning in a positive direction
When changing the potential scan direction at more negative potentials (Fig. 2, curve 6), the anodic maximum is split into two peaks corresponding to different alloy phases—possibly a solid solution of tin in silver and some other tin-richer phase. The corresponding diffraction spectrum of a coating deposited at −1,400 mV (Fig. 3, dashed line) shows the maxima of small amounts of tin and silver. The coating consists mainly of a Ag3Sn phase. In the corresponding anodic curve of dissolution (Fig. 4, curve 1), a peak of pure tin is still not registered, possibly due to its passivation or replacement by silver. However, β-tin reflexes remain in the diffraction spectrum after repetitive scans in the positive direction up to 0 V (Fig. 3, dotted line) and the disappearance of the split peak of the silver containing phases (Fig. 4, curve 2) is observed.

Anodic linear sweep voltammograms of Ag–Sn coating: 1 (——), deposited at a potential E = −1,400 mV, first scan; 2 (- - -), after repetitive scans in positive direction; 3 (...), in BE in absence of Ag, first scan
A dissolution peak of tin is registered when a coating deposited at a potential of −1,400 mV is dissolved in the same electrolyte in the absence of silver (Fig. 4, curve 3) which confirms that a replacement reaction of tin with silver takes place.
As expected, an increase in tin concentration in the electrolyte leads to an increase in the rate of tin deposition (Fig. 5). In spite of the possibly higher tin amount deposited, a tin dissolution maximum is still not observed. The changes in the anodic part of the CV curves are possibly connected with increased passivation of the tin richer phases compared to the case represented by curve 1.

Influence of CSn: (——) 15 g dm−3 Sn (BE); (- - -) 20 g dm−3 Sn (electrolyte 1); sweep rate 25 mV s−1
At other metal concentrations, but the same ratio of both metals in the electrolyte, some depolarization as well as some acceleration of the cathodic reactions is observed, as expected. However, significant changes in the ratio of the rates of the different anodic reactions are not registered (Fig. 6).

Influence of metal ion concentration at constant ratio (1:1) on the electrode processes: BE (——); electrolyte 2 (- - -); sweep rate 25 mV s−1
The effects of the tin salt used and of the alkalinity of the electrolyte are shown in Fig. 7. When using Sn2P2O7 (curve 2) instead of SnCl2 (curve 1) no changes in the silver deposition reaction are observed, but in the absence of KOH (curve 2) which influences tin deposition more strongly than that of silver, some depolarization of tin deposition, even at higher concentrations of pyrophosphate ions, occurs (compared to curve 1).

Influence of tin (II) salt type and KCN amount on the electrode processes: electrolyte 1 (——); electrolyte 3 (- - -); electrolyte 4 (...); sweep rate 25 mV s−1
The alloy dissolution reaction is also well expressed due to the contribution of K4P2O7. An increase in the concentration of free KCN (curve 3) causes polarization of the cathodic reactions and additional depolarization of the anodic reaction due to the strong complex forming effect of KCN on silver.
The investigations show that the electrodeposition of the alloy from this type of electrolyte and the formation of different alloy phases are possible. Due to its passivation or replacement of tin with silver, dissolution of pure tin phase cannot be detected in the anodic branch of the voltammograms.
The complex forming agent EDTA affects the first cathodic reaction of silver deposition more strongly compared to K4P2O7. The effect of both complex forming agents on the second cathodic reaction is similar (Fig. 8). Their combination exerts a weak synergetic effect leading to additional inhibition of the first cathodic reaction which reflects in the diminished anodic maximum in the presence of both substances. An electrolyte containing a combination of both complex forming agents should have better exploitation parameters.

Effect of complexing agents Na2EDTA and K4P2O7 on silver deposition in the absence of Sn: curve 1 (——) BE; curve 2 (- - -) BE in the absence of K4P2O7; curve 3 (…….) BE in the absence of Na2EDTA; sweep rate 25 mV s−1
The deposition of coatings with up to 40 wt% tin is possible from the investigated electrolytes (Fig. 9). Tin percentage was measured on a Hull cell (25 cm3) sheet covered with the alloy with a current of 100 mA for 10 min. The percentage values were measured in the middle height of the covered area. The system is of regular type according to Brenner [3], so tin percentage increases with increasing current density up to about 1.5 A cm−2. There are no further changes at higher current densities because of diffusion limitations for the deposition of both metals.

Effect of current density on alloy composition. Hull plate from electrolyte 1
The possibility for electrodeposition of alloy coatings with significant amounts of tin which surpasses the saturation limit of the silver lattice with this metal and, therefore, the limit for the formation of a solid solution of tin in silver, allows the deposition of coatings with phase heterogeneity. In such a case the question about the spatial distribution of the different phases in the coating and on its surface arises. Figure 10 shows a heterogeneous coating (in the middle part of the Hull cell cathode) deposited at current densities between 0.8 and 1.0 A dm−2, with a tin content between 35 and 40 wt%. Self-organization phenomena, like waves and periodic spatio-temporal structures consisting of phases with different tin content were observed on the electrode surface; these are very similar to the structures observed during electrodeposition of other silver alloys, such as Ag–Sb [41–43], Ag–Bi [22] and Ag–In [32, 33]. Investigations of the changes in the scale and in the period of the observed spatio-temporal structures depending on the electrolysis conditions and the composition of the alloy coating are still in progress.

Surface morphology of an Ag–Sn coating deposited in the Hull cell from electrolyte 1. Current density region between 0.8 and 1.0 A cm−2; Sn content in the deposit about 35–40 wt%
4 Conclusions
The deposition of Ag–Sn coatings with up to 40–50 wt% Sn is possible from the investigated electrolytes. XRD results show the presence of Ag, Sn and Ag3Sn phases in the coatings. Phase heterogeneity is observed at high tin content in the coatings and results in ordered spatio-temporal structures similar to the structures observed in the electrodeposition of other silver alloys.
References
Malishev V, Rumjantzev D (1976) Serebro. Metalurgija, Moskva (russ.)
Ruemmler WP (1951) Process of plating bright silver alloy. US Patent 2,555,375, 5 June 1951
Brenner A (1963) Electrodeposition of alloys. Principles and practice, 1st edn, vol 2. Academic Press, New York and London
Fedotjev NP, Vjacheslavov PM (1970) Plating 7:700
Vjacheslavov PM, Grilihes SJ, Burkat GK, Kruglova EG (1970) Galvanotehnika blagorodnih i redkih metallov. Mashinostroenie, Leningrad
Raub E, Dehoust G, Ramcke K (1968) Metall 22:573
Kristev I, Nikolova M (1986) J Appl Electrochem 16:867
Grunwald E, Varhelyi C (1979) Galvanotechnik 70:437
Krastev I, Petkova N, Zielonka A (2002) J Appl Electrochem 32:811
Krastev I, Zielonka A (2002) J Appl Electrochem 32:1141
Izbekova OW, Kudra OK, Gaewskaja LW (1971) Ukr Khim Zhurn (russ.) 37:657
Brenner A (1963) Electrodeposition of alloys. Principles and practice, 1st edn, vol 1. Academic Press, New York and London
Vasquez SM, Riesgo O, Duffo GS (1997) Rev Metalurgia 33:311
Reksc W, Mrugalski T (1990) Plat Surf Finish 77:60
Aguf MI, Karbasov BG, Tikhonov KI (1988) Sov Electrochem 24:1296
Izbekova OW, Kudra OK, Gaewskaja LW (1971) Ukr Khim Zhurn 37:86
Tsertsvadze SI, Tkemaladze TS (1981) Electrolyte for precipitating silver-cadmium alloy coatings. SU840209, 23 June 1981
Tsertsvadze SI, Dodeliya MA (1981) Electrolyte for precipitating silver-cadmium alloy coatings. SU840210, 23 June 1981
Raub E, Wullhorst B (1947) Z Metallkunde 2:33
Raub E, Engel A (1950) Z Metallkunde 41:485
Krastev I, Valkova T, Zielonka A (2003) J Appl Electrochem 33:1199
Krastev I, Valkova T, Zielonka A (2004) J Appl Electrochem 34:79
Krastev I, Valkova T, Zielonka A (2005) J Appl Electrochem 35:539
Krastev I, Valkova T, Zielonka A (2005) Galvanotechnik 8:1790
Krastev I (1994) Metalloberfläche 48:319
Pushnapavam M, Shenoi BA (1977) Finish Ind March 17
Raub E, Engel A (1943) Z Elektrochem 49:89
Raub E (1938) Mitteilungen des Forschungsinstituts und Probieramts für Edelmetalle. Schwäbisch Gmünd 12:55
Raub E, Schall A (1938) Z Metallcunde 30:149
Daniel G (1931) Process of electrodepositing indium and indium and silver. US Patent 1935630, 21 Nov 1933
Krastev I, Baumgärtner ME, Raub ChJ (1997) Galvanotechnik 88:1882
Dobrovolska Ts, Veleva L, Krastev I, Zielonka A (2005) J Electrochem Soc 152:C137
Dobrovolska Ts, Krastev I, Zielonka A (2005) J Appl Electrochem 35:1245
Dobrovolska Ts, Krastev I, Zielonka A (2004) Galvanotechnik 95:872
Dobrovolska Ts, Krastev I, Zielonka A (2004) Galvanotechnik 95:1134
Grekova NA, Vjacheslavov PM (1971) Zh Prikl Khim 44:1975 (Russ.)
Kondo T, Obata K, Takeuchi T et al (1998) Plat Surf Finish, February:51
Nutsch R, Liebscher H (1992) Galvanotechnik 83:2586
Puippe J Cl, Fluehmann W (1983) Plat Surf Finish, January:46
Raub E, Muller K (1967) Fundamentals of metal deposition. Elsevier Publishing Company, Amsterdam, London, New York
Kristev I, Nikolova M (1986) J Appl Electrochem 16:875
Kristev I, Nikolova M, Nakada I (1989) Electrochim Acta 34:1219
Krastev I, Koper MTM (1995) Phyica A 213:199
John WG, Evans EI (1937) Philos Mag 23:1033
Dornblatt AJ (1940) In: Addicks L (ed) Silver in the industry. Reinhold Publishing Corporation, New York
Buckner SJ, Falk AH (1925) Solder. US Patent 1,565,115, 8 Dec 1925
Leach RH (1934) Alloy. US Patent 1,952,082, 27 March 1934
DU PONT (1939) Process for coating metals. DE Patent 670403, 18 Jan 1939
Leidheiser H Jr, Ghuman ARP (1973) J Electrochem Soc 120:484
Kubota N, Horikoshi T, Sato E (1984) Met Finish March:53
Kubota N, Sato E (1985) Electrochim Acta 30:305
Nutsch R, Liebscher H, Degner W et al (1993) Galvanotechnik 84:425
Arai S, Watanabe T (1998) Mater Trans. JIM 39:439
Watanabe T (2004) Nanoplating, microstructure control theory of plated film and data base of plated film microstructure. Elsevier Science Ltd, Amsterdam–Boston–Heidelberg–London–New York–Oxford-Paris–San Diego–San Francisco–Singapore–Sydney–Tokyo
Kim JY, Yu J, Lee JH et al (2004) J Electronic Mat 33:1459
Dietterle M, Jordan M (2005) Electrolyte and method for depositing tin-silver alloy layers. US Patent 20050029112A1, 10 Feb 2005
Herklotz G, Frey T (1996) Electroplating bath for the electrodeposition of silver-tin alloys. US Patent 5514261, 7 May 1996
Molodchadskij A, Vishomirskis R (1969) In: Yu M (ed) Blestjashtie electroliticheskie pokritija, Mintis,Viljnus
Acknowledgements
The authors thank Deutsche Forschungsgemeinschaft (DFG) for the support of project 436 BUL 113/97/0-4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hrussanova, A., Krastev, I. Electrodeposition of silver–tin alloys from pyrophosphate-cyanide electrolytes. J Appl Electrochem 39, 989–994 (2009). https://doi.org/10.1007/s10800-008-9746-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-008-9746-5