
Abstract
Commercial activated carbon (Norit R3ex), de-mineralised with conc. HF and HCl, was oxidised (conc. HNO3) and heat-treated at various temperatures (180, 300 and 420 °C). The physicochemical properties of the samples obtained were characterised by selective neutralisation and pH-metric titration of surface functional groups (acid–base properties), thermogravimetry (thermal stability—TG), FTIR spectroscopy (chemical structure) and low-temperature nitrogen adsorption (BET surface area). Thermal treatment of the carbon materials caused the surface functional groups to decompose; in consequence, the chemical properties of the carbon surfaces changed. Cyclic voltammetric studies were carried out on all samples using a powdered activated carbon electrode (PACE) and a carbon paste electrode (CPE), as were electrochemical measurements in aqueous electrolyte solutions (0.1 M HNO3 or NaNO3) in the presence of Cu2+ ions acting as a depolariser. The shapes of the cyclic voltammograms varied according to the form of the electrodes (powder or paste) and to the changes in the surface chemical structure of the carbons. The electrochemical behaviour of the carbons depended on the presence of oxygen-containing surface functional groups. The peak potentials and their charge for the redox reactions of copper ions \( \left( {{\text{Cu}}^{2 + } \leftrightarrow {\text{Cu}}^{ + } \leftrightarrow {\text{Cu}}^{0} } \right) \) depended on their interaction with the carbon surface.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Because of their well-developed internal surfaces and high reactivity, activated carbons are used as adsorbents for numerous technological, environmental and analytical purposes, including the removal of heavy metal ions from aqueous solutions [1, 2]. An understanding of the phenomena taking place when solutions containing metal ions come into contact with a carbon surface is important for the control of sorption/desorption processes, for acquiring knowledge of the catalytic properties of carbon materials with adsorbed metal species, and for explaining the electrochemical behaviour of carbon electrodes in an electrolyte solution. Generally speaking, these processes depend on the cationic species to be adsorbed, the adsorbent’s porosity and surface chemistry, and the conditions of adsorption. Numerous interactions can take place between metal ions and a carbon surface, e.g. surface complex formation and/or charge-transfer reactions [3]. Since they are widely used in electroanalysis, electrosynthesis, electrosorption and electrocatalysis, electrodes produced from various carbon materials have been extensively investigated, with emphasis on the factors affecting capacitance and faradaic currents [4–6]. Several different types of carbon electrode materials have been employed in such studies, including pyrographite [7], glassy carbon [8–12], carbon fibres [13], carbon paste [14–16] and carbon powder [3, 17]. Heavy metal ions in aqueous solutions are very often used as polarisers: metal and metal complexes are immobilised on the surface of carbon electrodes and other carbon supports in order to preconcentrate them from dilute solution [17], to investigate the reactions of the surface redox system [3, 12], and for catalytic [18] and electrocatalytic purposes [19–22].
The present work had three objectives: (i) to compare the electrochemical behaviour of carbon materials in relation to the type of electrodes (powder or paste) prepared from them; (ii) to assess the utility of these electrodes for investigating the physicochemical properties of activated carbon surfaces (the effects of chemical modification); and (iii) to acquire an understanding of the processes taking place when heavy metal ions in solution come into contact with an activated carbon surface.
2 Experimental
2.1 Materials
The commercial activated carbon R4ex (Norit, The Netherlands) was demineralised with conc. HF and HCl (ash content 0.2% wt.), then oxidised with conc. HNO3 at 355 K. The oxidised activated carbon was divided into three samples, each of which was heat-treated for 4 h under vacuum at a different temperature—180, 300 and 420 °C. The samples were respectively denoted 180, 300 and 420.
2.2 Acid–base properties
The chemical properties of the modified carbon surface were evaluated by standard neutralisation titration with aq. HCl and aq. NaOH (both 0.1 M). The pHs of 1 g carbon slurries in 100 cm3 0.1 M NaCl were also measured.
2.3 Direct pH-metric titration
Direct pH-metric titrations were done using an automatic set (Mettler—Toledo DL67) with a glass electrode (Mettler DGT-111SC) and a 5 mL burette. The electrolyte (0.1 M NaOH or HCl) was dosed at 0.04 μL s−1 into a vessel containing 100 mg carbon and 40 mL doubly distilled water.
2.4 Thermogravimetry
Thermogravimetric analysis was done on a Simultaneous TGA-DTA Thermoanalyser (SDT 2960—TA Instruments). The mass loss in the 150–600 °C temperature range was determined from the TG curves recorded in a helium atmosphere.
2.5 FT-IR spectroscopy
FT-IR spectra of the carbon samples were obtained using a Perkin-Elmer FTIR Spectrum 2000 spectrometer. Activated carbon-KBr mixtures (1:300) were ground in an agate mortar, desorbed under vacuum (10−2 Pa), and then hydraulically pressed into pellets. Prior to the recording of a spectrum, the background line was obtained arbitrarily and subtracted. The spectra were then recorded from 4,000 to 450 cm−1 at a scan rate of 0.2 cm s−1; the number of interferograms at a nominal resolution of 4 cm−1 was fixed at 25.
2.6 Surface area
The specific surface areas (S bet ) of the carbon samples were determined from low-temperature (77.4 K) nitrogen adsorption/desorption isotherms (ASAP-2010—Micromeritics). Micropore (Vmi) and mesopore (Vme) volumes were also calculated from the adsorption isotherms [23].
2.7 Electrochemical studies
Cyclic voltammetric measurements were performed using an Autolab (Eco Chemie) modular electrochemical system equipped with a PGSTAT 10 potentiostat, driven by GPES3 software and the typical three-electrode system. A powdered activated carbon electrode (PACE) or carbon paste electrode (CPE), Pt wire and saturated calomel electrode (SCE) were used as working, counter and reference electrodes respectively.
The working PACE design was the same as that used and described earlier [3, 17]. After prior vacuum desorption (10−2 Pa), the powdered carbon (grain size 0.075–0.060 mm, mass 50 mg) was placed in an electrode container and drenched with a de-aerated solution to obtain a 3–5 mm sedimentation layer. The potentiometric responses (Est) of the carbon electrodes were measured in oxygen-free electrolyte solution once their values had stabilised (usually after 24 h). The potential measurements were accurate to ±2 mV. Cyclic voltammetric curves (CVs) were recorded for selected sweep amplitudes following the establishment of electrochemical equilibrium (no changes in repeated CV scans).
The working CPE design was similar to the one described earlier [24]. The paste was made from powdered activated carbon (250 mg) and mineral oil (20 drops of Nujol), and a portion was packed into a Teflon holder (thickness of layer—5 mm, surface area—0.2 cm2). When electrochemical equilibrium was reached (no further shape changes in repeated scans), CV curves were recorded for selected sweep amplitudes.
Acidic (0.1 M HNO3) or neutral (0.1 M NaNO3) solutions were used as blank electrolytes; copper (Cu2+) in 0.05 M nitrate solution was applied as depolariser. The sweep parameters used (rates, amplitudes) are given each time in the Figure captions.
3 Results and discussion
Oxidation of the carbon samples with nitric acid and their subsequent thermal treatment altered their surface chemistry. There was a significant (ca. 5-fold) decrease in acidity with a parallel increase in basicity (Table 1). Accordingly, the pH of the carbon slurry in neutral solution increased with the heat treatment temperature of the oxidised carbon. Titrations using a pH-meter were done with NaOH (a) and HCl (b) solutions (both 0.1 M) (Fig. 1a, b, respectively). The NaOH solution dosage range corresponded to the quantity of neutralised acidic surface groups decomposing during thermal treatment below 420 °C. These are mainly carboxylic groups in various environments on the carbon surface. The other groups were thermally more stable in these conditions. As the heat treatment temperature increased, the curves approached the level of the blank electrolytes. The number of acidic groups decreased as their acidic strength did so. The neutralisation of the basic groups was almost complete within the titrant (HCl) dose range.

Direct pH-metric titration curves with NaOH (a) and HCl (b) solutions for modified carbon samples: 1—180, 2—300, 3—420; dashed line—blank solution
Table 1 lists the mass losses, determined from the TG curves, of the carbon samples in the 150–600 °C temperature range. Mass loss decreased considerably with increasing HTT. According to the literature [1], thermal treatment reduced the quantity of acidic functional surface groups (mainly carboxyl) decomposing with the evolution of CO2.
Such significant changes in surface chemistry were accompanied by relatively small changes in porous structure (Table 2). The observed increase (ca. 10%) in specific surface area determined using the multipoint BET method (SBET) and micropore volume (Vmi) was typical of heat-treated carbon samples previously oxidised with conc. HNO3 [3]. During oxidation, some of the micropores became blocked by the surface oxygen complexes that formed; these were decomposed by heat treatment and the pores reopened.
Next, the FT-IR studies of the changes in the carbon samples caused by heat-treatment were carried out. The broad, overlapping band of stretching OH vibrations (maximum at 3,430 cm−1) in the FTIR spectra (Fig. 2) of the carbon materials was due to surface hydroxylic groups in different surroundings and to chemisorbed water. The asymmetry of this band at lower wave numbers indicated the presence of strong hydrogen bonds. Below 2,000 cm−1, the spectra showed absorption typical of a carbon surface and structural oxygen species. The spectra resembled the reported spectra of other carbons derived from a wide variety of sources. The presence of bands at 1,720 and 1,700 cm−1 can be respectively attributed to the stretching vibrations of C=O moieties in carboxylic and lactonic functional groups, and the changes in the absolute and relative intensities of these bands during thermal treatment at rising temperature can be explained by the disappearance of carboxylic structures and surface dehydration.

FT-IR spectra of the modified carbon samples
The overlapping bands near 1,575 cm−1 are due to ion-radical structures, conjugated systems such as diketone, keto-esters, keto-enol structures, and/or aromatic carbon [3]. Since H2O is sorbed on the surface of activated carbons with the participation of interactions both specific (hydrogen bonds, chemisorption due to surface oxide hydration) and non-specific (physical adsorption), the bands in the 1,500–1,600 cm−1 region may also be distorted by OH bending vibrations. The complicated nature of the adsorption bands in the 1,650–1,500 cm−1 region suggests that aromatic ring band and double bond (C=C) vibrations overlap the aforesaid C=O stretching vibration bands and OH binding vibration bands [3].
The partially resolved peaks forming the absorption band in the 1,260–1,000 cm−1 region (maxima at 1,185 and 1,114 cm−1) can be assigned to ether-like (symmetrical stretching vibrations), epoxide and phenolic (vibrations at 1,180 cm−1) structures existing in different structural environments. The changes in the relative intensities of these bands during thermal treatment at rising temperatures suggest that the number of ether and hydroxylic structures on the carbons investigated decreases, though to different degrees; this applies mostly to ether-like moieties [3].
Despite the changes in the surface chemistry, the differences in the porous structure were insignificant—witness the small differences in the low-temperature nitrogen adsorption isotherms (Fig. 3). Calculated specific surface areas (SBET) increased by only ca. 10% following the thermal treatment of the oxidised sample (Table 2), and the micropore volume (Vmi) changed in a similar manner. Variations in the mesopore volume (Vme) could be neglected, as they were over ten times smaller than the micropore volume.

Nitrogen adsorption–desorption isotherms at 77.4 K on the modified carbon samples (open symbols—desorption)
The electrochemical behaviour (in CV experiments) of electrodes prepared from the test carbons (powdered—PACE and paste—CPE for comparison) was investigated at potential ranges from −0.2 to +1.1 V vs. SCE (PACE) or from −0.2 to +0.9 V vs. SCE (CPE) and for sweep rates of 0.002 V s−1 (PACE) or 0.04 V s−1 (CPE). Only capacitive currents were visible on the CV curves for all samples (Fig. 4—PACE; Fig. 5—CPE) in both acidic (0.1 M HNO3) and neutral (0.1 M NaNO3) blank solutions. The potential responses (Est) for PACEs in acid and neutral environments depended on the degree of carbon surface oxidation and decreased markedly with rising thermal treatment temperature (Table 3). The electric double layer capacity for PACE (Cdl*), determined by extrapolation to the zero potential sweep rate, also decreased with the degree of carbon surface oxidation, but the changes in capacity were far smaller than the acid–base uptake (see Table 1). Similar results were obtained for powdered electrodes prepared from activated carbon thermally treated over a wide temperature range [5, 25]. For CPE the decrease in the electric double layer capacity (Cdl**) was more pronounced (especially in an acidic environment) during the thermal decomposition of the surface oxides. CV curves recorded for the carbon 180 CP electrode in acidic solution exhibited not only capacitive currents but also ‘pseudocapacitive’ or faradaic currents. The electric double layer capacity increased with electrode surface oxidation for the glassy carbon electrode [10]; this can be explained by the redox reactions of surface quinone/hydroquinone systems, but it is more probably due to the uptake or release of hydrogen ions [10].

Cyclic voltammograms recorded in aq. 0.1 M HNO3 (a) and aq. 0.1 M NaNO3 (b) for PACE prepared from the modified carbon samples

Cyclic voltammograms recorded in aq. 0.1 M HNO3 (a) and aq. 0.1 M NaNO3 (b) for CPE (in Nujol) prepared from the modified carbon samples
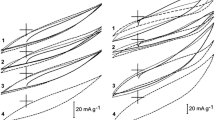
The addition of Cu2+ as depolariser to the blank solutions caused the stabilised potential response of the PACEs (Est in Tables 4, 5) to increase by 50–100 mV, the values being higher in an acidic environment. At the same time, however, the stabilised shapes of the CVs changed significantly, and only faradaic currents were recorded. One pair of anodic/cathodic peaks is present in all systems (Fig. 6a, b); the anodic peak is well shaped, whereas the cathodic process appears to take place in two stages (two overlapping peaks). As suggested by different authors [25, 26], the formation of a metallic Cu layer on the electrode surface implies two consecutive charge-transfer steps involving the formation of a Cu+ intermediate: a two-electron cathodic reduction, or two-one-electron cathodic reductions of adsorbed copper ions [27, 28]. The cathodic response according to the scheme:
may be responsible for the peaks present on the CV curves recorded in acidic (Fig. 6a) and neutral (Fig. 6b) electrolyte solution containing 0.1 M Cu(NO3)2. Thus, a well-shaped anodic peak represents a process that is the reverse of the one in Scheme 1. Tables 4 and 5 list the peak potentials determined from CVs recorded for particular electrochemical systems. The differences in the potential values can be explained by changes in the surface chemistry of the carbon materials. For the carbon 180 samples in an acidic medium, the next reaction may be explained by ion-changing adsorption (Schemes 3 and 4) and a possible surface redox reaction with electrochemically active functional groups (Scheme 5):

Cyclic voltammograms recorded for PACE in acidic (a) and neutral (b) aqueous solution containing Cu2+ ions (0.1 M Cu(NO3)2). Sweep rate: 2 mV s−1
The adsorbed species can then take part in the redox process shown in Schemes 1 and 2. Although the anodic part of the CV curves exhibits a well-shaped oxidation peak, the cathodic part displays a broad reduction wave. Gradual removal of surface oxides (carbon samples 300 and 420) causes a decrease in the anodic peak potential (Table 4) and a significant increase in the cathodic peak current (Fig. 6a). For carbon 420, the cathodic reaction (broad reduction wave) probably proceeds mainly according to Scheme 2.
The CVs recorded for CPEs in acid and neutral electrolyte solutions containing copper ions (Fig. 7a, b respectively) also exhibit differences in relation to the curves recorded for PACEs, namely, a pair of cathodic and anodic peaks, the potentials of which are dependent on the carbon modification procedure and the electrolyte’s pH. Polarisation of the CPE at negative potentials leads directly to the reduction of Cu2+ ions in a two-stage process according to the scheme
and is clearer cut than in case of the PACE, especially in neutral solution.

Cyclic voltammograms recorded for PCE in acidic (a) and neutral (b) aqueous solution containing Cu2+ ions (0.1 M Cu(NO3)2). Sweep rate: 40 mV s−1
Tables 4 and 5 give the estimated peak potentials for acidic and neutral media respectively. In the absence of any complexing anions, Cu(I) is unstable and undergoes disproportionation. From our results, we can quantitatively represent the oxidation of the Cu0 species in an acidic medium by:
Cu+ then disproportionates:
We suggest the following reaction to explain peak a2:
The higher anodic peak (a1) observed for all carbon samples in acidic (especially) and neutral media indicates that, after electrochemical oxidation of the adsorbed copper(II) ions, disproportionation of Cu(I) ions occurred in accordance with Scheme 8. The peak current increased with the gradual removal of surface oxides. Tables 6 and 7 list the differences in charge values (calculated as peak areas) for acidic and neutral electrolyte solutions respectively. The changes in peak charge values to a large extent confirm our supposition about Cu2+ ions redox processes occurring on the surface of the carbon electrodes.
4 Conclusion
Thermal treatment of the carbon materials altered the chemical properties of their surfaces. The shapes of the cyclic voltammograms varied in relation to changes in the surface chemical structure of the carbons when copper ions were used as depolarisers on powdered activated carbon and carbon paste electrodes. The electrochemical behaviour of the carbons tested in redox reactions depended on the presence of oxygen-containing surface functional groups, and the peak potentials of the redox reactions of adsorbed copper species \( \left( {{\text{Cu}}^{2 + } - {\text{Cu}}^{ + } - {\text{Cu}}^{0} } \right) \) depended on the interaction of these species with the carbon surface.
There were distinct differences in the electrochemical behaviours of electrodes made from the same carbon material in various forms (sedimentation layer or paste in Nujol). This may be explained by the dominant influence of the adsorption equilibrium of the depolariser species on the carbon material in the case of the powdered electrode. In the case of the paste electrode, however, diffusion-controlled processes played the main part.
References
Bansal RC, Goyal M (2005) Activated carbon adsorption. CRC/Taylor and Francis Group, Boca Raton
Radovic LR, Moreno-Castilla C, Rivera-Utrilla J (2001) In: Radovic LR (ed) Chemistry & physics of carbon, vol 27. Marcel Dekker, Inc, New York, p 227
Biniak S, Świątkowski A, Pakuła M (2001) In: Radovic LR (ed) Chemistry & physics of carbon, vol 27. Marcel Dekker, Inc, New York, p 125
Kinoshita K (1988) Carbon: electrochemical and physicochemical properties. Wiley, New York
Hsieh CT, Teng H (2002) Carbon 40:667
Pakula M, Biniak S, Swiatkowski A, Neffe S (2002) Carbon 40:1873
Beilby AL, Carlsson A (1988) J Electroanal Chem 448:283
Surya A, Murthy NA (1993) Electroanalysis 5:265
Yang Y, Lin ZG (1994) J Electroanal Chem 364:2
Shin H, Park M, Kim AR, Kang C (2003) J Electroanal Chem 547:143
Drissi-Daoudi R, Irhzo A, Darchen A (2003) J Appl Electrochem 33:339
Cofre P, Bustos A (1994) J Appl Electrochem 24:564
Jannakoudakis AD, Jannakoudakis PB, Theodoridou E, Besenhard JO (1990) J Appl Electrochem 20:619
Kalcher K, Kauffmann JM, Wang J, Svancara I, Vytras K, Neuhold C, Yang Z (1995) Electroanalysis 7:5
Darlewski W, Swiatkowski A (1997) Przemysł Chemiczny 76:440 (in Polish)
Lopes da Silva WT, Thobie-Gautier C, Rezende MOO, El Murr N (2002) Electroanalysis 14:71
Pakula M, Swiatkowski A, Biniak S (1995) J Appl Electrochem 25:1038
Shiu KK, Shi K (1998) Electroanalysis 10:959
Dandekar A, Baker RTK, Vannice MA (1999) J Catal 183:131
Pletcher D, Sheridan AJ (1998) Electrochim Acta 43:3105
Jannakoudakis AD, Jannakoudakis PB, Pagalos N, Theodoridou E (1994) Electrochim Acta 39:1881
Garjonyte R, Malinauskas A (1998) Sens Actuators B Chem 46:236
Jankowska H, Świątkowski A, Choma J (1991) Active carbon. Ellis Horwood, New York
McCreery RL, Cline KK (1996) In: Kissinger PT, Heineman WR (eds) Laboratory technique in electroanalytical chemistry, 2nd edn. Marcel Dekker, New York
Guaus E, Torrent-Burgues J (2007) Portugaliae Electrochim Acta 25:139
Driad JP, Montella C (2006) J Electroanal Chem 590:126
Bond AM (1980) Modern polarographic methods in analytical chemistry. Marcel Dekker, New York
Biniak S, Pakuła M, Szamański GS, Świątkowski A (1999) Langmuir 15:6117
Acknowledgements
This work was partly supported by the Polish Ministry of Science and Higher Education (project No. 3 T09B 067 29).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Biniak, S., Pakuła, M., Darlewski, W. et al. Powdered activated carbon and carbon paste electrodes: comparison of electrochemical behaviour. J Appl Electrochem 39, 593–600 (2009). https://doi.org/10.1007/s10800-008-9697-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-008-9697-x