
Abstract
The electrooxidation of benzylamine, 2-phenylethylamine and 4-hydroxyphenylethylamine (tyramine) at a gold electrode in contact with an alkaline electrolyte solution was studied. The effect of amine concentration, electrolyte pH and potential scan rate on the electrooxidation was analysed. The adsorption of amines on the gold-solution interface was found to play a significant role in the oxidation mechanism. The rate determining step was the heterogeneous dehydrogenation of the amine molecule, involving electron transfer to the gold electrode and the formation of a water molecule. The catalytic effect of the gold electrode on both benzylamine and 2-phenylethylamine oxidation is higher than that for tyramine.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
A detailed knowledge of the electrochemical behaviour of organic compounds is of primary importance for development of appropriate sensors and for advanced electrochemical synthesis. Numerous studies deal with electrocatalytic oxidation of organics on noble metals. An effective catalyst in electrochemical oxidation of organic substances should have the following properties: (i) the energy of adsorption of substrate molecules and/or intermediate species formed on the catalysts surface should be sufficiently high to substantially decrease the activation energy for the dehydrogenation step, but sufficiently low for the intermediate and final products to desorb easily, and (ii) the energy released by formation of water molecules as one of the reaction products should be sufficient to compensate for the energy needed for desorption of dehydrogenated organic intermediates from the metal surface. Among noble metals gold and platinum are the most active and most often used electrocatalysts. Gold stands out because of its good electrocatalytic activity in alkaline environments [1–14], whereas platinum is the most active in acid [15, 16]. Oxidation of organic substances at platinum is accompanied by a characteristic, though very undesirable, effect of surface blocking by strongly adsorbed intermediate species [16]. This effect does not apply to gold which has no vacancy in its d-bands and thus is characterized by relatively poor adsorption properties [17–24] as compared with platinum. Though these adsorption properties of gold account for its weak catalytic activity in acid solution, in alkaline solutions, when hydroxide ions adsorb on the gold surface [12, 25–34], gold proves to be a more effective catalyst for dehydrogenation of organic compounds than platinum.
The organic compounds selected for the study presented here are aliphatic amines. They have been widely used as starting or intermediate agents in the production of various other chemical compounds. Amines have found application in different branches of industry, agriculture and pharmacology [35, 36]. Most of them are toxic; thus their monitoring and detection are of great significance in environmental protection [35]. The literature gives rather scarce experimental evidence for the electrochemical behaviour of aliphatic amines. For gold, it has been established that oxidation of ethylamine in alkaline media occurs concomitantly with gold oxide formation [37–40]. More recently, relationships between the adsorption properties and electrocatalytic oxidation of a homologous series of aliphatic amines ranging from methylamine to butylamine have been evaluated and discussed [41–46]. The effect of molecular structure on isomeric butylamines has been analysed [47].
This article deals with the electrochemical behaviour of aliphatic amines with aromatic substituent: benzylamine, 2-phenylethylamine and 4-hydroxyphenylethylamine (tyramine) at the gold electrode in aqueous solution. Phenyletylamines (derivatives of 2-phenylethylamine) exist in biological systems as alkaloids and hormones [35, 36]. They play an important role in brain chemistry because some of them are neurotransmitters in the mammalian central nervous system [48]. Thus better understanding of different properties of these amines is of particular importance from both fundamental and practical points of view.
2 Experimental
Electrochemical studies were performed in a conventional three-electrode cell using a gold bead (99.999% purity) of 0.3 cm2 geometric area as a working electrode and a gold sheet as a counter-electrode. A hydrogen electrode (RHE) in the supporting electrolyte solution as proposed by Will et al. [49, 50] was used as a reference electrode. For comparison with published data, all the potentials are given versus the saturated calomel electrode (SCE).
The solutions were prepared using water purified in a Millipore Milli-Q system, benzylamine, 2-phenylethylamine and 4-hydroxyphenylethylamine (tyramine) of Fluka, NaOH and NaClO4 of Merck. All chemicals were analytical reagent grade and were used without further purification. The electrochemical measurements were carried out at 295 K. All solutions under investigation were deaerated with high purity argon.
The apparatus used to record cyclic voltammograms (j–E curves) and differential capacitance of the electrical double layer versus electrode potential curves (C–E) was described earlier [6]. The C–E curves were obtained by superimposing an ac sinusoidal voltage signal (15 s−1, 0.005 V) on a slow linear electrode potential scan (dE/dt = 0.005 V s−1). A slower scan rate of 0.002 V s−1 did not affect the C–E curves observed. Stirring of the solutions had no effect on the course of the C–E curves, which indicated that the mass transport did not limit the rate of adsorption–desorption processes of amine at the bulk concentrations studied. The equilibrium of the adsorption–desorption was checked at different frequencies. For frequencies in the range 5–40 s−1 no frequency dispersion of the differential capacity was observed in the potential range −0.95 V < E < 0.1 V and −0.95 V < E < 0.05 V in 0.1 mol dm−3 NaOH and 0.02 mol dm−3 NaClO4, respectively. These potential ranges narrow in the presence of amines in the supporting electrolyte to −0.4 V < E < 0.1 V in NaOH and to −0.4 V < E < 0.05 V in NaClO4 at all the adsorbates concentrations studied. These findings confirm that the differential capacity data in the above-mentioned potential range can be considered at equilibrium.
Prior to each series of measurements with amines, the working electrode was electrochemically activated by cycling (dE/dt = 0.1 V s−1) in the potential range between where no faradaic reactions occur on the gold surface. This procedure avoids structural changes on the gold surface.
3 Results and discussion
Comprehensive recognition of the adsorption phenomena at electrified interfaces is of the utmost importance for understanding of the mechanisms of reactions in electrocatalysis. The representative set of the C–E curves in 0.1 mol dm−3 NaOH as supporting electrolyte and at different concentration of benzylamine, phenylethylamine and tyramine shown in Fig. 1 demonstrates the adsorption of amines under investigation.

Differential capacitance versus potential for the gold electrode in 0.1 mol dm−3 NaOH (curve 1–10) with increasing benzylamine (▲) concentration; (2) 0.00001 mol dm−3; (3) 0.005 mol dm−3; (4) 0.05 mol dm−3 and with increasing phenylethylamine (■) concentration; (5) 0.00001 mol dm−3; (6) 0.005 mol dm−3; (7) 0.035 mol dm−3 and with increasing tyramine (●) concentration; (8) 0.00001 mol dm−3; (9) 0.005 mol dm−3; (10) 0.02 mol dm−3. dE/dt = 0.005 V s−1
A gradual suppression of the capacitance curves with increasing bulk amine concentration in the electrolyte takes place in the potential range of the characteristic minimum in the vicinity of the potential of zero charge (E pzc) of the gold electrode [51–54] and of the adsorption–desorption maximum on the negatively charged surface. This behaviour is clear evidence for the substitution of water with amine molecules at the gold surface. Relatively strong adsorption of the solute on gold is manifested by the fact that the C–E curves for the highest amine concentrations do not merge at the negative potential limit with the corresponding C–E curve for supporting electrolyte solution. This proves that some adsorbate molecules remain at the gold surface.
Analysis of relationships between the surface coverage, Θ = (C Θ = 0 − C Θ)/(C Θ = 0 − C Θ = 1) (where: C Θ = 0, C Θ = 1 and C Θ represent differential capacitance of the double layer at Θ = 0, Θ = 1 and at intermediate coverages (0 < Θ < 1), respectively) and the amine concentration, c A suggests increasingly stronger interaction of organic molecules with the gold electrode in the following sequence: benzyloamine < phenylethylamine < tyramine. The surface coverage for the same bulk amine concentration at chosen E is the highest for tyramine (Fig. 2). The concentration responsible for Θ = 1 decreases in the same order. The minimum differential capacitance (C Θ = 1) is reached at 0.05 mol dm−3, at 0.035 mol dm−3 and at 0.02 mol dm−3 for benzylamine, phenylethylamine and for tyramine, respectively.

Dependence of the gold electrode coverage on the amine concentrations in 0.1 mol dm−3 NaOH at E = −0.25 V versus SCE for: benzylamine (▲), phenylethylamine (■) and tyramine (●). dE/dt = 0.005 V s−1
It should be emphasised that the potential range in which the amine molecules are adsorbed on the gold-alkaline solution interface coincides with that in which the adsorption of hydroxyl ions occurs [12, 20, 27, 28, 55]. Thus a quantitative evaluation of the C–E curves in this medium is not possible because of the competing adsorption of amine and hydroxyl ions at the electrode surface and only qualitative information about the relative adsorptivity of the system can be obtained. Therefore, additional measurements of differential capacitance with a non-adsorbing supporting electrolyte, 0.02 mol dm−3 NaClO4 [51], have been made. The results reveal that the potential range of the amine adsorption, as measured with respect to E pzc, does not change with electrolyte pH.
It has been established that the dependence of the surface coverage (Θ) on the bulk amine concentration in the proximity of the potential of zero charge of the gold electrode can be represented by the Frumkin isotherm \( [\Uptheta /(1 - \Uptheta )]\exp ^{ - 2a\Uptheta } = \beta c \) [56] where: “a” stands for the lateral interaction parameter and β is the adsorption equilibrium constant. Figure 3 shows the \( [\ln c - \ln \Uptheta /(1 - \Uptheta )] \) vs. Θ isotherm plot resulting from analysis of the C–E curves at the potential of maximum adsorption, E max = −0.3 V versus SCE determined from the relationships between \( \Updelta C = C_{\Uptheta = 0} - C_\Uptheta \) versus E for given solutes concentration.

Test of the Frumkin isotherm for amines adsorbed at E max = −0.3 V versus SCE on gold electrode in 0.02 mol dm−3 NaClO4 for: benzylamine (▲), phenylethylamine (■) and tyramine (●). dE/dt = 0.005 V s−1
Taking as the reference state the unit mole fraction of amine in the bulk of the solution and a monolayer coverage (Θ = 1) of the non-interacting adsorbate molecules at the surface [21, 24, 57–60], β is related to the standard Gibbs energy of adsorption \( \Updelta G_{{\text{ad}}}^0 \) by the following relationship: \( \beta = [(1/55.5)\exp ( - \Updelta G_{{\text{ad}}}^0 /RT)] \). The corresponding values of \( \Updelta G_{{\text{ad}}}^0 \) change in the sequence: benzylamine (\( \Updelta G_{{\text{ad}}}^0 \) = −40.0 ± 0.05 kJ mol−1 [61]) <phenylethylamine (\( \Updelta G_{{\text{ad}}}^0 \) = −42.7 ± 0.04 kJ mol−1) <tyramine (\( \Updelta G_{{\text{ad}}}^0 \) = −46.0 ± 0.06 kJ mol−1), reflecting the stronger interaction between the amine molecule and the electrode surface. The amine molecules are able to form surface complexes by coordinative interaction between the electrode surface and the electron lone pair at the nitrogen atom and tyramine is moreover able to interact with gold by lone electron pairs localized at the oxygen atom.
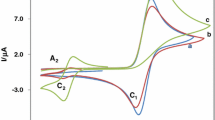
Figure 4 shows the cyclic voltammograms recorded at the gold electrode in the supporting electrolyte, 0.1 mol dm−3 NaOH alone, and in the presence of 0.005 mol dm−3 benzylamine, phenylethylamine and tyramine. A well-defined anodic peak at E = 0.33 V and a cathodic peak at E = 0.15 V visible in the j–E curve of the bare electrode are assigned to the gold oxide formation and to its reduction, respectively [55]. As follows from the cyclic voltammograms, aliphatic amines are inactive at the gold electrode potentials below E ≈ −0.1 V. Their oxidation proceeds irreversibly only in parallel with gold oxide formation and the anodic peaks with maxima at E = 0.43 V, E = 0.38 V and E = 0.45 V appear on the cyclic voltammograms for benzylamine, phenylethylamine and tyramine, respectively.

Cyclic voltammograms of gold electrode in 0.1 mol dm−3 NaOH (x) and with 0.005 mol dm−3 of: benzylamine (▲); phenylethylamine (■) and tyramine (●). dE/dt = 0.1 V s−1
When comparing the response of the gold electrode in supporting electrolyte solution with those in the presence of amines, it is evident that the charge used for the reduction of gold oxide in the presence of amines is smaller than that in solution without solute. This points to participation of gold oxide in the oxidation of amines. The same conclusion was drawn for aliphatic amines without aromatic substituent [45–47] and for methanol oxidation on the polycrystalline gold electrode [52, 62].
For all the amines studied a characteristic feature is an increase in the anodic current density with increasing concentration. As an example, the cyclic voltammograms recorded in the presence of benzylamine of concentration ranging from 0.0001 mol dm−3 to 0.005 mol dm−3 are presented in Fig. 5. In the positive scan of potential the maximum in the oxidation peaks shifts positively when the bulk amine concentration increases. Such a shift is typical of reactions involving molecules adsorbed on the electrode surface. An increase in the amine coverage generates a decrease in the gold oxide coverage. This conclusion is supported by the fact that any increase in the anodic peak is accompanied by a decrease in the cathodic peak.

Cyclic voltammograms of gold electrode in 0.1 mol dm−3 NaOH (x) and with benzylamine: 0.0001 mol dm−3 of (▲); 0.001 mol dm−3 (■) and 0.005 mol dm−3 (●). dE/dt = 0.1 V s−1
The similarity in the shape of the cyclic voltammograms for the compounds investigated is a result of the similarity of their molecular structure. An increase in the carbon chain length of the organic molecule (benzylamine and phenylethylamine) is accompanied by an increase in the electrochemical activity of the amine. This effect is significantly higher when compared with that observed for methylamine and ethylamine [45, 46]. This may be a consequence of the greater standard Gibbs energy of adsorption for amines with an aromatic ring then for those without such a substituent [41–44]. The smaller current density observed for tyramine, having the same carbon chain as phenylethylamine, must be a consequence of the presence of the hydroxyl group in the aromatic ring of the tyramine molecule which is able to interact with the gold surface besides the NH2 group. This possibility may be responsible for blocking the electrode surface for further substrate adsorption and oxidation. The low level fouling of the electrode surface by tyramine molecules was confirmed in a separate experiment in which the electrode was cycled in solution with this amine, then rinsed and dipped in fresh supporting electrolyte. The resulting voltammograms showed a tyramine oxidation peak, which disappeared after the third scan. Similar measurements with both benzylamine and phenylethylamine did not show any traces of them at the electrode after transfer to pure electrolyte.
For all amines studied the oxidation peak potential shifts positively with increasing sweep rate, confirming the irreversibility of the electrocatalytic process. The log j p versus log v plots (Fig. 6) are linear with an average slope of \( {\text{d}}\log j_{\text{p}} /{\text{d}}\log v \) of 0.81 ± 0.01, 0.82 ± 0.01 and 0.71 ± 0.01 for benzylamine, phenylethylamine and tyramine, respectively. The magnitudes of this parameter may indicate a mixed, i.e. diffusion-adsorption rate control [63].

The logarithm of peak current density versus the logarithm of potential sweep rate for benzylamine of (▲) 0.001 mol dm−3 and (△) 0.01 mol dm−3; for phenylethylamine of (■) 0.001 mol dm−3 and (□) 0.01 mol dm−3 and for tyramine of (●) 0.001 mol dm−3 and (ο) 0.01 mol dm−3 on gold electrode in 0.1 mol dm−3 NaOH
In order to get information on the rate determining step of amine oxidation, Tafel analysis of voltammograms was made. The slopes, \( {\text{d}}E/{\text{d}}\log j = {\text{2}}{\text{.303}}RT/\alpha nF \) of the linear parts of the plots in log j–E coordinates (Fig. 7) in the potential range −0.05 V and 0.2 V for both benzylamine and phenylethylamine and between 0.1 V and 0.3 V for tyramine give values of 0.270 ± 0.002 V, 0.260 ± 0.003 and 0.210 ± 0.003 V per decade of current density for 0.001 mol dm−3 benzylamine, phenylethylamine and tyramine, respectively. The change in potential sweep rate did not alter the Tafel slopes significantly. Moreover, the dE/log j slopes increase somewhat with increasing amine concentration. The electron transfer coefficient, α, derived from the Tafel slopes, lower than 0.5 for all the amines examined, support the assumption of an adsorption step preceding the first electron transfer step determining the overall reaction rate. Further steps proceed so fast that they have no influence on the rate of the process considered.

Dependence of current density on gold electrode potential in 0.1 mol dm−3 NaOH for: 0.001 mol dm−3 benzylamine: dE/dt = 0.1 V s−1 (▲) and dE/dt = 0.01 V s−1 (△); for 0.001 mol dm−3 phenylethylamine: dE/dt = 0.1 V s−1 (■) and dE/dt = 0.01 V s−1 (□) and for 0.001 mol dm−3 tyramine: dE/dt = 0.1 V s−1 (●) and dE/dt = 0.01 V s−1 (○)
The decisive role of the adsorption steps in the kinetics of aliphatic amine electrooxidation on gold is indicated by the fractional reaction order with respect to the substrate concentration, z A. Figure 8 displays the plots of the logarithm of current density versus the logarithm of substrate concentration at a constant electrode potential of 0.05 V for benzylamine and phenylethylamine and of 0.2 V for tyramine. The slope of the straight lines of the appropriate log j–log c A relationships gives the reaction order, \( z_{\text{A}} = {\text{d}}j/{\text{d}}c_{\text{A}} \) which in the concentration range between 0.0005 mol dm−3 and 0.02 mol dm−3 is equal to 0.22 ± 0.01 for benzylamine, 0.20 ± 0.01 for phenylethylamine and 0.20 ± 0.02 for tyramine. z A increases insignificantly with decreasing potential sweep rate (Fig. 9).

Dependence of the current density on the amine concentrations in 0.1 mol dm−3 NaOH at E = 0.05 V versus SCE for benzylamine: dE/dt = 0.1 V s−1 (▲) and dE/dt = 0.01 V s−1 (△); at E = 0.05 V versus SCE for phenylethylamine: dE/dt = 0.1 V s−1 (■) and dE/dt = 0.01 V s−1 (□) and at E = 0.2 V versus SCE for tyramine: dE/dt = 0.1 V s−1 (●) and dE/dt = 0.01 V s−1 (○)

Dependence of peak potential on pH of the supporting electrolyte solution for 0.001 mol dm−3: benzylamine: dE/dt = 0.1 V s−1 (▲) and dE/dt = 0.01 V s−1 (△); phenylethylamine: dE/dt = 0.1 V s−1 (■) and dE/dt = 0.01 V s−1 (□) and tyramine: dE/dt = 0.1 V s−1 (●) and dE/dt = 0.01 V s−1 (ο)
Finally, analysis of the variation of the peak potential E p with electrolyte pH shows that during the electrode process, not only electrons, but also protons are released from the organic molecules. The peak potentials shift linearly to less positive potentials with increasing pH. The linear regression obtained from pH = 11 to pH = 13.3 showed a slope close to 0.06 V per unit pH for all the amines. The magnitude of the \( {\text{d}}E_{\text{p}} /{\text{dpH}} \) slope suggests that the number of electrons transferred is equal to that of protons taking part in the electrooxidation reaction.
Thus the possible reaction pathways for benzylamine, phenylethylamine and tyramine oxidation on gold in alkaline solution can be proposed as shown in Scheme 1. The possible reaction pathways for benzylamine and phenylethylamine oxidation (Scheme 1) involve the first electron transfer connected with the oxidation of the hydrogen atom abstracted from the NH2 group of an adsorbed amine molecule as the reaction rate determining step. The resulting imines are then either oxidized to the respective nitriles or, after imine hydrolysis, ammonium and the respective aldehydes may be produced. The subsequent oxidation of the latter products to the respective acids can be ruled out because in such a case the value of \( {\text{d}}E_{\text{p}} /{\text{dpH}} \) should be different from 0.06 V per unit pH. The same conclusion holds for tyramine. However, for tyramine, the -OH group linked to the aromatic ring is irreversibly oxidized before the oxidation of the NH2 group. As a result the iminoketone is formed. This blocks the electrode surface which, besides stronger interactions with the metal, is probably the reason for the lowest current density obtained on gold during tyramine oxidation as compared with those observed for benzylamine and phenylethylamine.

Possible reaction pathways for electrooxidation of benzylamine, phenylethylamine and tyramine on a gold electrode in an alkaline solution
References
Bełtowska-Brzezinska M, Vielstich W (1977) Electrochim Acta 22:1313
Bełtowska-Brzezinska M (1979) Electrochim Acta 24:409
Bełtowska-Brzezinska M (1980) Electrochim Acta 25:267
Bełtowska-Brzezinska M (1980) Kinetics and mechanism of electrocatalytic oxidation of alcohols on gold, platinum and gold–platinum alloys, edited by UAM Press, Poznań
Beden B, Cetin I, Kahyaoglu A, Takky D, Lamy C (1987) J Catal 104:37
Holze R, Łuczak T, Bełtowska-Brzezinska M (1990) Electrochim Acta 35:1345
Beden B, Leger JM, Lamy C (1992) In: O`M Bockris J, Conway BE, White RE (eds) Modern Aspects of Electrochemistry, vol 22. Plenum Press, New York, pp 97–264 and references therein
Łuczak T, Bełtowska-Brzezinska M, Holze R (1993) J Appl Electrochem 3:1039
Holze R, Łuczak T, Bełtowska-Brzezinska M (1994) Electrochim Acta 39:485
Holze R, Łuczak T, Bełtowska-Brzezinska M (1994) Electrochim Acta 39:991
Łuczak T, Holze R, Bełtowska-Brzezinska M (1994) Electroanalysis 6:773
Bełtowska-Brzezinska M, Łuczak T, Holze R (1997) J Appl Electrochem 27:999
Bełtowska-Brzezinska M, Łuczak T (2001) Electroanalysis 15:1270
Borkowska Z, Tymosiak-Zielinska A, Shul G (2004) Electrochim Acta 49: 1209
Raichewa SN, Kalcheva SV, Christov MV, Sokolova EI (1974) J Electroanal Chem 55:213
Parsons R, VanderNoot T (1988) J Electroanal Chem 257:9
Wieckowski A (1975) J Electrochem Soc 122:252
Bełtowska-Brzezinska M, Dutkiewicz E, Skołuda P (1984) J Electroanal Chem 181:235
Bełtowska-Brzezinska M, Heitbaum J (1985) J Electroanal Chem 183:167
Holze R, Bełtowska-Brzezinska M (1986) J Electroanal Chem 201:387
Lipkowski J, Stolberg L (1992) In: Lipkowski J, Ross PN (eds) Adsorption of molecules at metal electrodes, vol. 1. Wiley-VCH, New York
Trasatti S (1992) Electrochim Acta 37:2137
Łuczak T, Bełtowska-Brzezinska M, Holze R (1993) Electrochim Acta 38:717
Bełtowska-Brzezinska M, Łuczak T, Holze R (1998) Surf Sci 418:281
Hauffe W, Heitbaum J (1978) Ber Bunsenges Phys Chem 82:487
Takky D, Beden B, Léger JM, Lamy C (1983) J Electroanal Chem 145:461
Desilvestro J, Weaver MJ (1986) J Electroanal Chem 209:377
Ocon P, Alonso C, Celdran R, Gonzales-Velasko J (1986) J Electroanal Chem 206:179
Burke LD, Lee BH (1992) J Electroanal Chem 330:637
Burke LD, Buckley DT, Morrissey JA (1994) Analyst 119:841
Burke LD, Moran JM, Nugent PF (2003) J Solid State Electrochem 7:529
Angerstein-Kozlowska H, Conway BE, Burnett B, Mozota J (1979) J Electroanal Chem 100:417
Adzić RR, Markocić NM, Vesović VB (1986) J Electroanal Chem 165:532
Alonso C, Gonzáles-Velasco J (1988) J Electroanal Chem 248:193
Morrison RT, Boyd RN (1985) Organic chemistry. PWN, Warsaw
Pawełczyk E (1986) Chemistry of medicines. PZWL, Warsaw
Luo P, Zhang F, Baldwin RP (1991) Anal Chim Acta 244:169
Jackson WA, Lacourse WR, Dobberpuhl DA, Johnson DC (1991) Electroanalysis 3:607
Dobberpuhl DA, Johnson DC (1995) Anal Chem 67:1254
Dobberpuhl DA, Johnson DC (1996) Electroanalysis 8–9:726
Łuczak T (2005) Collect Czech Chem Commun 70:2027
Łuczak T (2005) Annals of the Polish Chemical Society 1:600
Łuczak T (2006) Coll Surf A 280:125
Łuczak T (2006) Asian Chem Lett 10:25
Łuczak T (2006) Collect Czech Chem Commun 71:1371
Łuczak T (2007) J Appl Electrochem 37:461
Łuczak T (2007) J Appl Electrochem 37:269
Patric GL (2005) An introduction to medical chemistry. Oxford University Press Inc, New York, p 593
Will FG, Hess HJ (1973) J Electrochem Soc 120:1
Will FG (1986) J Electrochem Soc 133:454
Clavilier J, Nguyen Van Huong C (1977) J Electroanal Chem 80:101
Borkowska Z, Stimming U (1991) J Electroanal Chem 312:237
Hamelin A, Stoicoviciu L (1987) J Electroanal Chem 234:93
Kolb DM (1993) In: Lipkowski J, Ross PN (eds) Structure of electrified interfaces, VCH, New York, p 98
Newman RC, Burstein GT (1981) J Electroanal Chem 129:343
Damaskin BB, Petrii OA, Batrakov V (1975) Adsorption organischer Verbindungen an Elektroden. Akademie-Verlag, Berlin
Mohilner DM, Nakadomari H, Mohilner P (1977) J Phys Chem 81:244
Nikitas P (1984) J Electroanal Chem 170:333
Jastrzębska J, Jurkiewicz-Herbich M, Trasatti S (1987) J Electroanal Chem 216:21
Cholewa E, Burgess I, Kunze J, Lipkowski J (2004) J Solid State Electrochem 8:693
Łuczak T, Bełtowska-Brzezinska M, Born M, Holze R (1997) Vib Spectrosc 15:17
Avramov-Ivić M, Jovanović V, Vlajnić G, Popić J (1997) J Electroanal Chem 423:119
Kisza A (2001) Electrochemistry II. WNT, Warsaw, p 94
Acknowledgements
Financial support by the Ministry of Scientific Research and Information Technology, Poland, is gratefully acknowledged. The author thanks Professor M. Bełtowska-Brzezinska for valuable discussions and A. Wojciechowska for technical help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Łuczak, T. Electrochemical behaviour of benzylamine, 2-phenylethylamine and 4-hydroxyphenylethylamine at gold. A comparative study. J Appl Electrochem 38, 43–50 (2008). https://doi.org/10.1007/s10800-007-9396-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-007-9396-z