
Abstract
The immune system is essential for the control and elimination of infections, and macrophages are cells that act as important players in orchestrating the various parts of the inflammatory/immune response. Amino acids play important role in mediating functionality of the inflammatory response, especially mediating macrophages functions and cytokines production. We investigated the influence of glutamine, taurine and their association on the modulation of inflammatory pathway markers in macrophages. The RAW 264.7 macrophage cell line was cultivated in the presence of glutamine and taurine and proliferation rates, cell viability, cell cycle phases, IL-1α, IL-6, IL-10 and TNF-α as well as H2O2 production and the expression of the transcription factor, NFκB, and its inhibitor, IκBα, were evaluated. Our results showed an increase in viable cells and increased proliferation rates of cells treated with glutamine concentrations over 2 mM, as well as cells treated with both glutamine and taurine. The cell cycle showed a higher percentage of cells in the phases S, G2 and M when they were treated with 2 or 10 mM glutamine, or with glutamine and taurine in cells stimulated with lipopolysaccharide. The pNFκB/NFκB showed reduced ratio expression when cells were treated with 10 mM of glutamine or with glutamine in association with taurine. These conditions also resulted in reduced TNF-α, IL-1α and H2O2 production, and higher production of IL-10. These findings demonstrate that glutamine and taurine are able to modulate macrophages inflammatory pathways, and that taurine can potentiate the effects of glutamine, illustrating their immunomodulatory properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Macrophages are cells that have important immune functions, providing a first line of defence, engulfing bacteria and producing inflammatory mediators (Stow and Condon 2016). Among these inflammatory mediators are cytokines and reactive oxygen species (ROS), which play a central role in inflammatory cell signaling, modulating cellular activities and coordinating cell behavior (Gardiner and Mills 2016). Hydrogen peroxide (H2O2) is a ROS which presents important bactericidal and tumoricidal activities (Dale et al. 2008) as well as cytokines such as TNF-α, IL-1 and IL-6 are proinflammatory cytokines and all of these mediators are involved in the regulation of inflammatory reactions. Usually, proinflammatory cytokines are modulated by the activation of the transcription factor, nuclear factor kappa B (NFκB) (Fock et al. 2010; Nishanth et al. 2011; Arango Duque and Descoteaux 2014). On the other hand, IL-10 plays an important role in the control of the inflammatory response attenuating NFκB activation, being also regarded as an anti-inflammatory, immunosuppressive cytokine (Fock et al. 2008; Ma et al. 2015).
For many years, the potential physiological uses of nutrients have been studied, particularly for the purpose of improving or controlling the immune response, such as by modulating macrophage cells. Among the wide range of nutrients studied, glutamine and taurine can be highlighted (Ferreira 2007; Grimble 2005). Glutamine is a nonessential amino acid, but during stress, injury or illness is considered a conditionally essential nutrient (Lacey and Wilmore 1990). Glutamine is the most abundant amino acid in plasma and tissues, providing energy substrate and serving as a precursor to amino acid synthesis (Newsholme 2001; Rogero et al. 2008a). Additionally, a large number of cells utilize glutamine at high rates, especially immune cells, which means that glutamine has immunomodulatory potential and a diminished supply of glutamine to immunocompetent cells can affect their functions (Newsholme et al. 1996; Newsholme 2001; Rogero et al. 2008b). Glutamine is utilized at high rates by macrophages (Newsholme et al. 1996). Whose ability to phagocytose pathogens, kill fungi and produce IL-1, IL-6, TNF-α and reactive oxygen species are modulated by the extracellular concentration of glutamine (Yassad et al. 2000; Rogero et al. 2008c; Kim 2011).
Unlike glutamine, taurine is not incorporated into proteins, but does play a role in many important physiological functions, including bile acid conjugation, protein stabilization, retinal and neurological development, osmoregulation, calcium mobilization, homeostasis and immune function (Newsholme et al. 2003; Kim 2011; Moe-Byrne et al. 2016). In addition, several studies have demonstrated its important role as an antioxidant. In neutrophils, taurine attenuates the toxicity of hypochlorous acid (HOCl) produced by the myeloperoxidase system. By reacting with HOCl, taurine generates a stable compound that is less toxic to cells, known as taurine chloramine (TauCl) (Sun et al. 2012; Menzie et al. 2013; Kim and Cha 2014).
Considering that numerous advances have been made regarding the influence of specific nutrients on the immune system, this study was based on the hypothesis that glutamine, taurine and their association are able to act on the inflammatory response, especially by modulating macrophage activities.
Materials and methods
Cell culture
RAW 264.7 macrophage-like cells were purchased from the American Type Culture Collection [No TIB-71; American Type Culture Collection (ATCC), Manassas, VA, USA] and cultured in Dulbecco’s modified Eagle medium–high glucose media (DMEM-HG; Cultilab, Campinas, Brazil), supplemented with 10% fetal bovine serum, Hepes buffer (2 g/L) and 1% penicillin–streptomycin. The culture had a final pH between 7.2 and 7.4, and was incubated in 5% CO2 at 37 °C and 100% humidity until it was ∼ 70% confluent.
Cells were detached from the culture plates using a cell scraper, centrifuged at 1500 rpm for 5 min, then reseeded in four separate culture plates at a density of 1.0 × 106 cells/mL, in free bovine serum (FBS)-starved media (DMEM-HG with 1% penicillin–streptomycin only). After a 16-h, the cells were incubated for 24 h with 0, 0.6, 2 or 10 mmol/L of glutamine, or 0, 1, 5 or 10 mmol/L of taurine, or with both amino acids in the same concentrations. The cultured RAW cells were stimulated for 30 min or 24 h, depending on the experiment, with 1.25 µg/mL of LPS (Escherichia coli 055:B5; Sigma-Aldrich, St Louis, MO, USA). The concentrations of glutamine were chosen for the following reasons: 0 mmol/L was the negative control; 0.6 mmol/L of glutamine corresponds to the normal concentration found in the plasma of healthy humans and rodents; 2 mmol/L is the standard concentration that is commonly used in cell cultures (Eagle 1955; Eagle et al. 1956); 10 mmol/L was chosen because previous enterocyte and peripheral blood mononuclear cell studies have shown that glutamine concentrations varying from 2 to 10 mmol/L had opposing effects on the expression of cytokines encoding genes (Wischmeyer et al. 2003; Hubert-Buron et al. 2006). The concentrations of taurine were chosen for the following reasons: 0 mmol/L was the negative control; 1 mmol/L was used because in this dosage significantly reduces oxidative stress (Kim and Kim 2005); 5 mmol/L was used as an intermediate concentration; 10 mmol/L was chosen because the intracellular concentration of taurine in mammalian tissues is commonly over 10 mmol/L (Fukuda et al. 1982).
MTT assay
RAW 264.7 cells were harvested and cell proliferation was determined using the MTT-Tetrazolium (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide); Sigma Chemical Company, St. Louis, MO, USA) assay. Cells were plated in 96-well plates and cultured 24 h at a density of 1.0 × 104 cells, after the experimental periods 50 µL of an MTT colorimetric solution was added to the wells and incubated for 4 h. After this, 500 µL of 10% sodium dodecyl sulfate (SDS) was added to each well and leaved at room temperature in the dark for 2 h. The absorbance of each well was determined using an ELISA microplate reader at 570 nm. The standard samples and cell viability curves were analyzed. Duplicates of crescent cell dilutions were distributed in 96-well plates in culture medium as follows: blank, 1 × 103, 1 × 104, 1 × 105, 5 × 105, 7.5 × 105 and 1 × 106. Absorbance values were converted into cell concentrations using a logarithmic equation, and then an average line chart for the standards was plotted.
Cellular viability assay
RAW 264.7 cells were adjusted to a final concentration of 1 × 106, cultured as described previously and stimulated for 24 h with 1.25 µg/mL of LPS. After the experiments periods, cells were harvested in phosphate-buffered saline (PBS; Sigma Chemical Co., St. Louis, MO, USA), centrifuged and the pellet was resuspended with 50 μL of annexin buffer and incubated with 3 μL of annexin-V and 5 μL of propidium iodide (PI; Becton–Dickinson Pharmingen, San Diego, CA, USA) for 20 min, protected from light. After incubation, cells were centrifuged and resuspended in 200 μL of annexin buffer for data acquisition using a FACS Canto II flow cytometer. At least 1 × 104 cells were acquired. Data analysis was performed using FlowJo 7.6.4 (Tree Star, Ashland, USA) software.
Cell cycle evaluation
RAW 264.7 cells were cultured as described previously and stimulated for 24 h with 1.25 µg/mL of LPS. 1 × 106 RAW 264.7 cells were harvested and fixed with 70% ethanol for 20 min on ice. After centrifugation, the pellet of the cells was resuspended with 4 mg/mL RNase and 4 µL of PI for 45 min at 37 °C, protected from light. Data acquisition and analysis by flow cytometry was performed as described above. Cell cycle status was assessed by quantifying the percentage of histogram regions corresponding to G1/G0 and S/G2/M phases.
Cytokine and H2O2 production
RAW 264.7 cells were cultured and treated as previously described. Once confluent, cells were trypsinized and seeded into 6-well plates at a density of 1 × 106 cells per mL and stimulated with 1.25 µg/mL LPS for 24 h. RAW 264.7 cell supernatants were collected and used to determine the production of IL-1α, IL-1β, IL-6, IL-10, TNF-α and H2O2. Cytokine production was assessed by enzyme-linked immunosorbent assay (ELISA) using a commercially available kit (Quantikine ELISA®, R&D Systems, Abingdon, UK) and H2O2 was measured using H2O2 assay kit Amplex red® (A22188, Invitrogen, Carlsbad, CA, USA).
Western blot analysis
For the analyses of transcription factors RAW 264.7 cells were cultured as described previously and stimulated for 30 min with 1.25 µg/mL of LPS. Western blot analysis was used to establish protein expression of IκBα, phosphorylated IκBα, NF-κB and phosphorylated NF-κB. The RAW 264.7 cells were cultured and treated as previously described and were washed with PBS three times and lysed with RIPA buffer (0.1% SDS, 1% Igepal CA-630, 1% sodium deoxycholate, 10 mM Tris HCl [pH 7.5], 150 mM NaCl, 2 μg/mL aprotinin, 1 μg/mL leupeptin, 100 μg/mL PMSF and 0.5 mM EDTA). A protease and phosphatase inhibitor cocktail was added (Sigma Chemical Co., St. Louis, MO, USA) to inhibit the activity of proteases and phosphatases. After centrifugation at 14,000 rpm and 4 °C for 15 min, the supernatant was collected, mixed with 5× Laemmli buffer (1 M Tris HCl [pH 6.8], 10% 2-mercaptoethanol, 10% SDS, 50% glycerol and 0.01% bromophenol blue) and boiled for 5 min. The protein content of the cell homogenates was determined using a BCA Protein Assay Kit (Pierce Biotechnology, Inc., Rockford, IL, USA), and equal amounts of protein (18 μg per well) were separated on 10% SDS–polyacrylamide mini-gels and transferred to Immobilon polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA, USA). These were incubated with the appropriate primary antibodies (1:1000), including IKBα (cat no. 4812, Cell Signaling Technology, Inc., Beverly, MA, USA), phosphorylated IKBα (cat no. 9246, Cell Signaling Technology, Inc., Beverly, MA, USA), NFκB (cat no. pc137, Calbiochem, San Diego, CA, USA) and phosphorylated NFκB (cat no. 3031, Cell Signaling Technology, Inc., Beverly, MA, USA). The membranes were then incubated at room temperature overnight with the primary antibody and for 1 h with a secondary antibody (1:5000), conjugated to horseradish peroxidase (cat no. DC03L, Calbiochem, San Diego, CA, USA). After three washes with Tris-buffered saline, 0.1% Tween 20 (TBST), the immunoreactive bands were visualized using the ECL detection system (Amersham ECL™ Advance Western Blotting Detection Kit, Piscataway, NJ, USA). To standardize and quantify the immunoblots, a digital detection system (ImageQuant™ 400 version 1.0.0, Amersham Biosciences, Pittsburgh, PA, USA) was used. The results were expressed in relation to the intensity of β-actin (1:20,000 for anti-β-actin; Cell Signaling Technology, Inc., Beverly, MA, USA) and as a percentage of the control value.
Statistical analysis
After a normality test, datasets were analyzed using the Student’s t test and the level of significance adopted was 95% (p < 0.05). For multiple comparisons among groups, analysis of variance and Bonferroni’s post hoc test were performed. Values were expressed as mean ± standard deviation. Statistical analyses were performed using the GraphPad Prism® software version 5.01 (GraphPad Software Inc., La Jolla, USA).
Results
In vitro proliferation assay
The results show differences in proliferation rates depending on the treatment. Cells stimulated with LPS for 24 h and cultured in 10 mM of glutamine (Fig. 1a), as well as cells cultured with 2 mM of glutamine plus 5 mM of taurine or cells cultured with 10 mM of glutamine plus 10 mM of taurine (Fig. 1c), showed increased proliferation rates when compared to the negative control (0 mmol/L).

The results are expressed as the mean ± SD of the proliferation rates of RAW 264.7 cells cultivated with glutamine, taurine or in association with both amino acids, and stimulated or not wit LPS. In each amino acid concentration studied, six samples of each group were evaluated. Asterisk illustrates a significant (p ≤ 0.05) difference between the treatment group and the control group treated with 0 mmol/L
Cell viability
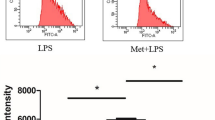
To test whether the amino acids affected cell viability, flow cytometry using annexin and propidium iodide was performed. An increase in cell viability was observed in cells stimulated with LPS and cultivated in the presence of 2 or 10 mM of glutamine (Fig. 2a). Cells cultivated with taurine did not show a difference in cell viability percentage when compared to cells cultivated without taurine (Fig. 2b). Cells cultivated with both amino acids showed an increase in cell viability when the cells were stimulated and cultivated with the two higher concentrations of the amino acids studied (Fig. 2c).

The results are expressed as the mean ± SD of the percentage of cell viability of RAW 264.7 cells cultivated with glutamine, taurine or in association with both amino acids, and stimulated or not with LPS. In each amino acid concentration studied, six samples of each group were evaluated. Asterisk illustrates a significant (p ≤ 0.05) difference between the treatment group and the control group treated with 0 mmol/L
Cell cycle
The cell cycle status of RAW 264.7 cells was evaluated in cells stimulated with LPS and cultivated in different concentrations of amino acids. RAW 264.7 cells cultivated with glutamine (2 and 10 mM) and stimulated with LPS resulted in a lower percentage of cells in the G0/G1 cell cycle phase when compared to the control (0 mM), due to a higher percentage of cells in the proliferative state (S/G2/M cell cycle phases; Fig. 3a). The experiments performed with taurine did not show differences among groups (Fig. 3b). Cells cultivated in the presence of both glutamine and taurine showed similar results to cells cultivated only with glutamine: the percentage of cells in the S/G2/M cell cycle phases was higher than cells cultivated without both amino acids (Fig. 3c).

The results are expressed as the mean ± SD of the percentage of cell cycle phases of RAW 264.7 cells cultivated with glutamine, taurine or in association with both amino acids, and stimulated or not with LPS. In each amino acid concentration studied, six samples of each group were evaluated. Significant differences between the treatment groups and the control group treated with 0 mmol/L are illustrated by *(p ≤ 0.05), **(p ≤ 0.01) and ***(p ≤ 0.001)
Cytokine and H2O2 production
TNF-α, IL-1α, IL-6 and IL-10 as well as H2O2 production were evaluated. Groups treated with 0.6, 2 and 10 mM glutamine and stimulated with LPS showed reduced production of TNF-α when compared to the control (0 mM of glutamine; Fig. 4a). In cells that were cultivated with taurine, statistical differences were not observed (Fig. 4b). However, cells treated with both amino acids and stimulated with LPS, and that received higher concentrations of glutamine and taurine (10/10 mM), showed reduced TNF-α production when compared to cells receiving 0/0 mM (Fig. 4c).

The results are expressed as the mean ± SD of the cytokines and H2O2 produced by RAW 264.7 cells cultivated with glutamine, taurine or in association with both amino acids, and stimulated or not with LPS. In each amino acid concentration studied, six samples of each group were evaluated. Asterisk illustrates a significant (p ≤ 0.05) difference between the treatment group and the control group treated with 0 mmol/L
The production of IL-1α showed similar results to those observed for TNF-α (Fig. 4d, f). However, reduced production of IL-1α was not observed when cells were cultivated with 0.6 mM of glutamine and stimulated with LPS, which were conditions that resulted in reduced production of TNF-α. Taurine did not induce alteration in the IL-1α production profile among groups (Fig. 4e). In addition, IL-6 production did not differ statistically among groups that were stimulated with LPS and cultivated with glutamine, taurine or both (Fig. 4g–i).
IL-10 is an anti-inflammatory cytokine and it produced opposite results to those for proinflammatory cytokines. Cells that received 10 mM of glutamine in the presence of LPS (Fig. 4j) showed an increase in production when compared to cells cultured without glutamine. Cells treated with 10 mM taurine and LPS had an increased production of IL-10 compared to the control group (0 mM; Fig. 4k). In the experiments using both amino acids, groups cultured with 2/5 and 10/10 mM plus LPS showed higher IL-10 production compared to the group that was not treated with these amino acids (0/0 mM; Fig. 4l).
There was a reduction in H2O2 production in cells treated with 10 mM of glutamine and stimulated with LPS in comparison to cells treated without glutamine or treated with 0.6 mM of glutamine (Fig. 4m). In cells cultivated with taurine, statistical differences were not observed (Fig. 4n). Cells treated with glutamine and taurine at concentrations 2/5 and 10/10 mM and stimulated with LPS showed reduced H2O2 production in comparison to cells treated without both amino acids (Fig. 4o).
NFκB and IKBα expression in RAW 264.7
Given that IKBα and NFκB have a central role in coordinating the inflammatory response, the expression of proteins was measured in RAW 264.7 cells treated with glutamine, taurine and their association. No statistical differences in protein expression were observed among groups without the LPS stimulus. For the RAW 264.7 cells stimulated with LPS, reduced expression of the ratio between phosphorylated and total NFκB when the cells were treated with 10 mM was observed in comparison to the control (0 mM of glutamine) (Fig. 5a). Treatment with taurine did not induce differences among groups stimulated with LPS. However, cells treated with both glutamine and taurine showed reduced expression of the pNFκB/NFκB ratio at concentrations of 2/5 and 10/10 mM (Fig. 5c). However, no statistical differences were observed in the ratio between phosphorylated and total IKBα among the groups stimulated with LPS and cultured with glutamine, taurine or both amino acids (Fig. 5c, d).

The results are expressed as the mean ± SD of pNFκB/NFκB ratio expression and IKBακB/NFκB ratio expression of RAW 264.7 cells cultivated with glutamine, taurine or in association with both amino acids, and stimulated or not with LPS. Results were represented in relation to the intensity of β-actin and are expressed in arbitrary units. In each amino acid concentration studied, six samples of each group were evaluated. Asterisk illustrates a significant (p ≤ 0.05) difference between the treatment group and the control group treated with 0 mmol/L
Discussion
Due to the importance of the immune system, many authors have investigated its activation, modulation and the specific mechanisms of how it can be improved. In this context, the use of some nutrients has also been applied for the purpose of intervening in and improving the immune system and the inflammatory response (Calder 2003; Grimble 2005). Among the most important nutrients for this purpose are amino acids, particularly glutamine and taurine.
In the present study, the cytotoxicity of the amino acids, glutamine and taurine, and their association in RAW 264.7 cells was analyzed after stimulation of the cells with LPS. The results demonstrated that, 10 mM of glutamine, preceded by the LPS stimulus, led to an increase in cell proliferation as well as cell viability when compared to cells cultured without glutamine. The same response occurred when the cells were cultured with glutamine in association with taurine, at concentrations of 2/5 and 10/10 mM. This was in contrast to cells cultured with taurine alone, which did not exhibit changes in these two parameters.
The proliferative status of the macrophage cells was confirmed by analysis of the cell cycle status. Cells cultured with 2 and 10 mM of glutamine, as well as cells cultured with glutamine in association with taurine, had a higher percentage of cells in the G2/S/M cell cycle phases when compared to negative glutamine control (0 mM). These results can be explained by the importance of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis which are biomolecules essential for cell proliferation (Ardawi and Newsholme 1983; Engström and Zetterberg 1984).
Glutamine participates in many biochemical pathways, including the synthesis of glutathione—a tripeptide that in the intracellular environment has a role as a co-factor for cytoplasmic enzymes and can regulate the cellular redox state (Ardawi and Newsholme 1983; Newsholme et al. 2003). Therefore, by being a precursor for glutathione synthesis, glutamine has an essential role in the cellular redox state, consequently acting in the modulation of sensitive enzymes and cell damage (Newsholme et al. 2003; Wischmeyer et al. 2003). At the same time, glutamine could modulate the activation of heat shock proteins (HSP), which have been correlated with repair and cellular defense (Wischmeyer et al. 1997, 2003). In addition, glutamine is a requirement for many cells in tissue culture, an intermediate in many metabolic pathways, and an alternative substrate to glucose for energy metabolism, suggesting that glutamine concentration might be a determinant of cell viability (Newsholme et al. 2003; Wischmeyer et al. 2003). In this way, the participation of glutamine in cellular repair is well known and may explain the increased cell viability found in cells treated with higher glutamine concentrations as observed in the current work.
In the literature, as already mentioned above, the proliferative effects of glutamine are already very well defined, however, in the opposite sense, the amino acid taurine, have been reported by some studies as apoptotic cellular inducer (Zhang et al. 2015). In the current study, taurine did not interfere in cell viability percentage when compared to cells cultivated without taurine. However, cells cultivated with both amino acids showed increased viability. There are studies that correlate both amino acids, where in multiple traumas or stress conditions, the plasma levels of taurine increased after the enteral diet was supplemented with glutamine (Boelens et al. 2003; Kim and Cha 2009). This suggests that glutamine substrate can act as a facilitator of the plasma availability of taurine and consequently improve the osmotic disturbances observed after trauma, and therefore, the physiological condition of these patients (Boelens et al. 2003; Kim and Cha 2009).
The macrophage activation by LPS occurs by binding to the Toll like receptor 4 (TLR-4), trigging the LPS/TLR-4 signal transduction pathway activating several transcription factors such as NFκB. The activity of NFκB is primarily regulated by interaction with inhibitory IKB proteins, specially IKBα (Gilmore 2006). In addition, the NFκB/IKBα interaction blocks the ability of NFκB to bind to DNA and results in the NFκB complex being primarily in the cytoplasm due to a strong nuclear export signal in IKBα (Gilmore 2006; Nishanth et al. 2011). After binding, some molecules involved with signal transduction and the phosphorylation of the inhibitor of NFκB, IκB-α, will be activated, resulting in IκB-α degradation and the subsequent release and migration of NFκB into the nucleus of the cell where it promotes the transcription of genes involved in the inflammatory response, such as genes encoding IL-1, IL-6 and TNF-α production (Fock et al. 2010; Nishanth et al. 2011). In the current study, there was a decrease in the p-NFκB/NFκB ratio when cells were cultured with 10 mM of glutamine or with glutamine in association with taurine, at concentrations of 2/5 and 10/10 mM. This data can be correlated with the alterations in cytokine production: a decrease in the proinflammatory cytokines, IL-1α and TNF-α, and an increase in the production of the anti-inflammatory cytokine, IL-10.
Moreover, in the current work was observed that glutamine in higher concentrations, in association or not with taurine, is able to reduce the macrophages H2O2 production. In this context, these results are in agreement with cytokines and NFκB results. H2O2 is a compound produced in a process known as the respiratory burst (Dale et al. 2008). H2O2 is a highly reactive compound produced by cells, especially macrophages, after secondary reactions during the course of inflammatory processes. H2O2 has an antimicrobicidal function but is also known that the H2O2 produced by the respiratory burst can act as a second messenger and activates major signaling pathways, showing that H2O2 directly activates NFκB and enhances the TNF-α production (Kim and Cha 2009; Karabay et al. 2015).
Our data showed that supraphysiological glutamine supplementation, especially 10 mM, are able to modulate proinflammatory markers and this results are in agreement with the literature, where it is reported that 4 mM of glutamine supplementation has the ability to decrease the production of TNF-α in mononuclear cells of peripheral blood, stimulated with LPS, and that in vivo supplementation with glutamine in free form or as a dipeptide decreases TNF-α release after intense exercise (Wischmeyer et al. 2003; Cruzat and Tirapegui 2009). Previous studies have shown that after multiple traumas or under stress conditions, there was an increase in the level of taurine in plasma after the enteral diet was supplemented with glutamine (Kim and Kim 2005; Miyazaki and Matsuzaki 2014). This suggests that glutamine acts as a substrate to taurine and facilitates the plasma availability of taurine, which can improve the osmotic disturbances commonly observed after traumas conditions (Boelens et al. 2003; Marcinkiewicz and Kontny 2014). This study suggests that glutamine and taurine are able to modulate macrophages inflammatory pathways, and that taurine can potentiate the effects of glutamine, illustrating their immunomodulatory properties. The effects shown here, relating to the improvement of cell viability, proliferation and the modulation of the inflammatory response, can be clinically exploited to improve the condition of patients.
References
Arango Duque G, Descoteaux A (2014) Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol 5:491. doi:10.3389/fimmu.2014.00491.eCollection
Ardawi MS, Newsholme EA (1983) Glutamine metabolism in lymphocytes of the rat. Biochem J 212:835–842. doi:10.1042/bj2120835
Boelens PG, Houdijk AP, de Thouars HN, Teerlink T, van Engeland MI, Haarman HJ, van Leeuwen PA (2003) Plasma taurine concentrations increase after enteral glutamine supplementation in trauma patients and stressed rats. Am J Clin Nutr 77:250–256
Calder PC (2003) Immunonutrition. BMJ 327:117–118
Cruzat VF, Tirapegui J (2009) Effects of oral supplementation with glutamine and alanyl-glutamine on glutamine, glutamate, and glutathione status in trained rats and subjected to long-duration exercise. Nutrition 25:428–435. doi:10.1016/j.nut.2008.09.014
Dale DC, Boxer L, Liles WC (2008) The phagocytes: neutrophils and monocytes. Blood 112:935–945. doi:10.1182/blood-2007-12-077917.Review
Eagle H (1955) The growth requirements of two mammalian cell lines in tissue culture. Trans Assoc Am Physicians 68:78–81
Eagle H, Oyama VI, Levy M, Horton CL, Fleischman R (1956) The growth response of mammalian cells in tissue culture to l-glutamine and l-glutamic acid. J Biol Chem 218:607–616
Engström W, Zetterberg A (1984) The relationship between purines, pyrimidines, nucleosides, and glutamine for fibroblast cell proliferation. J Cell Physiol 120:233–241. doi:10.1002/jcp.1041200218
Ferreira IK (2007) Nutritional therapy in intensive care unit. Rev Bras Ter Intensiva 19:90–97
Fock RA, Vinolo MA, Crisma AR, Nakajima K, Rogero MM, Borelli P (2008) Protein-energy malnutrition modifies the production of interleukin-10 in response to lipopolysaccharide (LPS) in a murine model. J Nutr Sci Vitaminol 54:371–377. doi:10.3177/jnsv.54.371
Fock RA, Rogero MM, Vinolo MA, Curi R, Borges MC, Borelli P (2010) Effects of protein-energy malnutrition on NF-kappaB signalling in murine peritoneal macrophages. Inflammation 33:101–109. doi:10.1007/s10753-009-9163-x
Fukuda K, Hirai Y, Yoshida H, Nakajima T, Usui T (1982) Free amino acid content of lymphocytes and granulocytes compared. Clin Chem 28:1758–1761
Gardiner CM, Mills KH (2016) The cells that mediate innate immune memory and their functional significance in inflammatory and infectious diseases. Semin Immunol 28:343–350. doi:10.1016/j.smim.2016.03.001
Gilmore TD (2006) NF-kB: from basic research to human disease. Oncogene (Reviews) 51:6679–6899
Grimble RF (2005) Immunonutrition. Curr Opin Gastroenterol 21:216–222
Hubert-Buron A, Leblond J, Jacquot A (2006) Glutamine pretreatment reduces IL-8 production in human intestinal epithelial cells by limiting IkappaB-alpha ubiquitination. J Nutr 136:1461–1465
Karabay AZ, Koc A, Gurkan-Alp AS, Buyukbingol Z, Buyukbingol E (2015) Inhibitory effects of indole α-lipoic acid derivatives on nitric oxide production in LPS/IFNγ activated RAW 264.7 macrophages. Cell Biochem Funct 33:121–127. doi:10.1002/cbf.3095
Kim H (2011) Glutamine as an immunonutrient. Yonsei Med J 52:892–897. doi:10.3349/ymj.2011.52.6.892
Kim C, Cha YN (2009) Production of reactive oxygen and nitrogen species in phagocytes is regulated by taurine chloramine. Adv Exp Med Biol 643:463–472. doi:10.1007/978-0-387-75681-3_48
Kim C, Cha YN (2014) Taurine chloramine produced from taurine under inflammation provides anti-inflammatory and cytoprotective effects. Amino Acids 46:89–100. doi:10.1007/s00726-013-1545-6
Kim JW, Kim C (2005) Inhibition of LPS-induced NO production by taurine chloramine in macrophages is mediated though Ras-ERK-NF-kappaB. Biochem Pharmacol 70:1352–1360. doi:10.1016/j.bcp.2005.08.006
Lacey JM, Wilmore DW (1990) Is glutamine a conditionally essential amino acid? Nutr Rev 48:297–309. doi:10.1111/j.1753-4887.1990.tb02967
Ma X, Yan W, Zheng H, Du Q, Zhang L, Ban Y, Li N, Wei F (2015) Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000Research 4. doi:10.12688/f1000research.7010.1 (pii: F1000 Faculty Rev-1465)
Marcinkiewicz J, Kontny E (2014) Taurine and inflammatory diseases. Amino Acids 46:7–20. doi:10.1007/s00726-012-1361-4
Menzie J, Prentice H, Wu JY (2013) Neuroprotective mechanisms of taurine against ischemic stroke. Brain Sci 3:877–907. doi:10.3390/brainsci3020877
Miyazaki T, Matsuzaki Y (2014) Taurine and liver diseases: a focus on the heterogeneous protective properties of taurine. Amino Acids 46:101–110. doi:10.1007/s00726-012-1381-0
Moe-Byrne T, Brown JV, McGuire W (2016) Glutamine supplementation to prevent morbidity and mortality in preterm infants. Cochrane Database Syst Rev 4:CD001457. doi:10.1002/14651858.CD001457
Newsholme P (2001) Why is l-glutamine metabolism important to cells of the immune system in health, post-injury, surgery or infection? J Nutr 131:2515S–2522S
Newsholme P, Costa Rosa LF, Newsholme EA, Curi R (1996) The importance of fuel metabolism to macrophage function. Cell Biochem Funct 14:1–10. doi:10.1002/cbf.644
Newsholme P, Procopio J, Lima MM, Pithon-Curi TC, Curi R (2003) Glutamine and glutamate—their central role in cell metabolism and function. Cell Biochem Funct 21:1–9. doi:10.1002/cbf.1003
Nishanth RP, Jyotsna RG, Schlager JJ, Hussain SM, Reddanna P (2011) Inflammatory responses of RAW 264.7 macrophages upon exposure to nanoparticles: role of ROS-NFκB signaling pathway. Nanotoxicology 5:502–516. doi:10.3109/17435390.2010.541604
Rogero MM, Borelli P, Fock RA, Pires ISO, Tirapegui J (2008a) Glutamine supplementation reverses impaired macrophage function resulting from early weaning in mice. Nutrition 24:589–598. doi:10.1016/j.nut.2008.02.005
Rogero MM, Borelli P, Vinolo M, Fock R, Pires I, Tirapegui J (2008b) Dietary glutamine supplementation affects macrophage function, hematopoiesis and nutritional status in early weaned mice. Clin Nutr 27:386–397. doi:10.1016/j.clnu.2008.03.004
Rogero MM, Tirapegui J, Vinolo MAR, Borges MC, Castro IA, Pires ISO et al (2008c) Dietary glutamine supplementation increases the function of peritoneal macrophages and hemopoiesis in early weaned mice inoculated with Mycobacterium bovis bacillus Calmette-Guérin. J Nutr 138:1343–1348
Stow JL, Condon ND (2016) The cell surface environment for pathogen recognition and entry. Clin Transl Immunol 5:e71. doi:10.1038/cti.2016.15
Sun K, Chen Y, Liang SY, Liu ZJ, Liao WY, Ou ZB, Tu B, Gong JP (2012) Effect of taurine on IRAK4 and NF-kappa B in Kupffer cells from rat liver grafts after ischemia-reperfusion injury. Am J Surg 204:389–395. doi:10.1016/j.amjsurg.2011.10.020
Wischmeyer PE, Musch MW, Madonna MB, Thisted R, Chang EB (1997) Glutamine protects intestinal epithelial cells: role of inducible HSP70. Am J Physiol 272:G879–G884
Wischmeyer PE, Riehm J, Singleton KD et al (2003) Glutamine attenuates tumor necrosis factor-alpha release and enhances heat shock protein 72 in human peripheral blood mononuclear cells. Nutrition 19:1–6. doi:10.1016/S0899-9007(02)00839-0
Yassad A, Husson A, Bion A, Lavoinne A (2000) Synthesis of interleukin 1beta and interleukin 6 by stimulated rat peritoneal macrophages: modulation by glutamine. Cytokine 12:1288–1291. doi:10.1006/cyto.1999.0729
Zhang X, Lu H, Wang Y, Liu C, Zhu W, Zheng S, Wan F (2015) Taurine induces the apoptosis of breast cancer cells by regulating apoptosis-related proteins of mitochondria. Int J Mol Med 35:218–226. doi:10.3892/ijmm.2014.2002
Acknowledgements
This work was supported by Fundação de Amparo a Pesquisa do Estado de São Paulo FAPESP (Grant no. 2016/16463-8). Rogero MM, Borelli P and Fock RA are fellows of the Conselho Nacional de Pesquisa e Tecnologia (CNPq).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sartori, T., Galvão dos Santos, G., Nogueira-Pedro, A. et al. Effects of glutamine, taurine and their association on inflammatory pathway markers in macrophages. Inflammopharmacol 26, 829–838 (2018). https://doi.org/10.1007/s10787-017-0406-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-017-0406-4