
Abstract
The present study was to investigate the therapeutical effects and mechanisms of Asiatic acid from Potentilla chinensis against alcoholic hepatitis. Rats were intragastrically fed with alcohol for 12 weeks to induce alcoholic hepatitis and then treated with various drugs for further 12 weeks. The results showed that Asiatic acid significantly alleviated liver injury caused by alcohol in rats, as evidenced by the improved histological changes and the lower levels of AST, ALT, and TBIL. Besides, Asiatic acid significantly enhanced the activity of ADH and ALDH, promoting alcohol metabolism. Asiatic acid suppressed CYP2E1 activity and NADP+/NADPH ratio, resulting in low ROS production. Further study revealed that Asiatic acid markedly reduced hepatocyte apoptosis by regulating the expression levels of apoptosis-related protein. Moreover, Asiatic acid could regulate the Nrf2 and NF-κB signaling pathway, attenuating oxidative stress and inflammation as a result. Interestingly, the comprehensive analysis of transcriptomics and metabolomics indicated that Asiatic acid inhibited the gene expression of Gpat3 and thereby affected the biosynthesis of the metabolites (1-acyl-Sn-glycerol-3-phosphocholine, phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine), regulating the glycerophospholipid metabolism pathway and ultimately ameliorating hepatocyte damage. In conclusion, this study demonstrates that Asiatic acid can ameliorate alcoholic hepatitis by modulating the NF-κB and Nrf2 signaling pathways and the glycerophospholipid metabolism pathway, which may be developed as a potential medicine for the treatment of alcoholic hepatitis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The liver is an important organ for drug metabolism and detoxification. Long-term overconsumption of alcohol may lead to the development of various alcohol-related liver diseases, including alcoholic fatty liver, alcoholic hepatitis, and alcohol‑related fibrosis [1]. Among alcoholic liver diseases, alcoholic hepatitis (AH) has 10 to 35% morbidity and few therapeutic options [2]. AH is characterized by jaundice and progressive inflammatory liver injury, closely associated with multiple metabolic alterations [3]. Chronic alcohol exposure can activate alcohol dehydrogenase and aldehyde dehydrogenase, resulting in overproduction of acetate and upregulation of cytochrome P450 2E1 (CYP2E1) expression, which induces alcoholic oxidative stress, pro-inflammatory mediators release, and fatty acid oxidation [4]. If alcoholic hepatitis disease isn’t suppressed, it is highly likely to develop into liver fibrosis, cirrhosis, and even liver cancer, for which there is currently no effective treatment [5]. Therefore, it is urgent to explore an effective therapy for AH.
Inflammatory cytokine production is mainly regulated by Nuclear factor-κB (NF-κB) activation, which subsequently promotes the transcription of numerous inflammation-associated genes, including IL-1β, IL‑6, and TNF-α, and provides the basis for the generation of these cytokines [6]. In addition, the oxidative stress induced by reactive oxygen species (ROS) plays an important role in the regulation of alcoholic hepatitis. A large amount of acetaldehyde and ROS are produced in the process of alcohol metabolism, and the excessive accumulation of ROS may trigger the oxidative stress response of the body [7]. Furthermore, many genes related to oxidative stress are regulated by the nuclear factor erythroid-2-related factor (Nrf2) [8], indicating that the NF-κB and Nrf2 signaling pathways may be the potential pathway to regulate inflammatory response and oxidative stress in alcoholic hepatitis.
In the last decade, metabolomics research has provided a unique advantage in identifying potential biological metabolic differences by analyzing metabolic changes caused by active substances [9, 10]. Metabolomics studies provide a framework for drug research by directly analyzing the phenotypic changes of drugs acting on tissues and elucidating their mechanisms [11]. Furthermore, transcriptomics has played a huge role in uncovering potential targets of disease. Hence, RNA-seq sequencing was used to observe the changes of mRNA and genes, and metabolomics based on UPLC-Q/TOF MS was applied to investigate the protective effect of AA on alcoholic hepatitis.
In our previous study, we found that Asiatic acid (AA) extracted from Potentilla chinensis had a strong anti-inflammatory and inhibitory effect on lipid accumulation [12]. However, whether AA has protective effects on AH remains unclear so far. In the present study, we performed integrated analyses of metabolomic and transcriptomic to reveal the underlying mechanism of AA against hepatitis. Glucocorticoids are often used as the final treatment for severe alcoholic hepatitis, but their side effects are obvious, which is not conducive to the long-term treatment of chronic diseases. Therefore, Silibinin as a natural liver-protecting drug, whose antioxidant effect is obvious, was selected as the positive control [13].
Materials and Methods
Ethanol-Induced AH Model in Rats
In our previous study, AA (purity > 98.0%) was isolated from P. chinensis [14]. Silibinin was purchased from Tianjin Tasly Shengte Pharmaceutical Co., Ltd. (Tianjin, China). A total of 60 Sprague Dawley (SD) male rats (Quality certificate number: SCXK Gui 2014–0002), weighing 160 ± 20 g were purchased from the Experimental Animal Center of Guangxi Medical University (Guangxi, China). All the procedures of animal care and experiment were approved by the Animal Ethics Committee of Guangxi Medical University.
The experiment schedule was shown in Fig. 1. Briefly, after 1 week of adaptive feeding, all the rats were randomly divided into six groups (10 rats per group): the normal group (I), AA control group (II), model group (III), Silibinin-treated group (IV), low-dosages of AA (V) and high-dosages of AA(VI) treatment groups. The therapeutic dosage of Silibinin and AA was chosen based on our previous study [12, 15]. Silibinin was dissolved in normal saline; AA was dissolved in 0.1% DMSO and then mixed in normal saline. The SD rats were given intragastric treatment once a day, respectively.

The experimental schedule
The ethanol dosages were given as follows: 30% ethanol 5.0 g/kg/d from 1 to 3 weeks, 35% ethanol 6.0 g/kg/d from 4 to 5 weeks, 40% ethanol 7.0 g/kg/d from 6 to 7 weeks, 40% ethanol 8.0 g/kg/d from 8 to 9 weeks, 40% ethanol 9.0 g/kg/d from 10 to 12 weeks, 40% ethanol 10.0 g/kg/d from 13 to 24 weeks. In addition to ethanol, rats inII, V, and VI groups were given AA; rats in the IV group were given Silibinin from 13 to 24 weeks daily. At the modeling period, the body weights of all rats were weighed every 2 days.
At the end of the treatment, all the rats were anesthetized and sacrificed, the blood and liver tissue was stored in the −80° refrigerator for further examination.
Histopathological and Hepatocyte Apoptosis Examination
A portion of liver tissues was prepared according to the previous studies [16]. The prepared slices were dried and dewaxed. Hematoxylin and eosin (H&E) staining were used for evaluating the pathological changes. The slices were stained with hematoxylin, washed with ultra-pure water, separated by hydrochloric acid ethanol, stained with eosin, dehydrated and transparent, and finally sealed with neutral glue.
Masson’s trichrome staining was used for evaluating hepatic collagen hyperplasia. The slices were stained with Weigert, washed with ethanol, stained with Masson cyanation and ponceau, washed with weak acid working fluid, stained with aniline blue, dehydrated and transparent, and finally sealed with neutral glue.
Besides, TUNEL staining was used to observe the apoptosis hepatocyte. The prepared slices were stained with 20 μg/mL protease K without DNase, washed with PBS, inactivated with 3% hydrogen peroxide solution, dehydrated and transparent, and finally sealed with neutral glue.
Biochemical Analysis
The blood of all rats was centrifuged, and then serum was collected. The serum activity of alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin (ALB), globulin (GLB), and total bilirubin (TBIL) was detected by an automatic biochemistry analyzer (Hitachi, Ltd., Kokubunji, Tokyo, Japan).
Assay for Lipid Levels
The levels of total cholesterol (TC), triglyceride (TG), high-density lipoprotein (HDL), and low-density lipoprotein (LDL) enzymes of liver tissues were detected by commercially available kits. The corresponding TC and TG reagents were added to the TC and TG plates respectively, then mixed and incubated in 37°for 10 min. Each well was measured the absorbance value at 510 nm wavelength.
The corresponding HDL and LDL reagents were added and mixed, respectively. The absorbance value at 510 nm wavelength of each well was measured immediately after 5 min of water bath. Then, the HDL and LDL plates were incubated in 37°for 10 min, measuring the absorbance value at 546 nm wavelength again.
Assay for Alcohol Metabolic Enzyme Activity
The liver tissues were homogenized in normal saline and centrifuged at 2500 × g at 4 °C for 10 min. The supernatants were measured ADH and ALDH activity by commercially available diagnostic kits. The experimental process strictly followed the instructions of the Alcohol dehydrogenase (ADH) assay kit and Aldehyde dehydrogenase (ALDH) assay kit. The corresponding ADH and ALDH reagent were added to the ADH and ALDH plates respectively, then mixed and incubated in 37°for 10 min. Distilled water and the sample were added (distilled water group as blank control). Each well was measured the absorbance value at 340 nm wavelength.
Determination of Oxidative Stress and Lipid Peroxidation
According to the manufacturer’s instruction, the above supernatants were used to detect the activities of superoxide dismutase (SOD), Glutathione peroxidase (GSH-Px), malonaldehyde (MDA), and myeloperoxidase (MPO) using the commercially available kits.
Total Superoxide Dismutase (T-SOD) assay kit was required setting up the determination tube and the opposite tube (adding distilled water in the opposite care and samples to the determination tube) and then adding the same reagent to fully mix. After incubation at 37° for 40 min, the chromogenic agent was added and the absorbance value of SOD in each well was measured at 550 nm wavelength.
Glutathione Peroxidase (GSH-Px) assay kit was required enzyme tubes (sample group), non-enzyme tubes (control group), blank tubes (distilled water group), and standard tubes (standard substance group) to be set, and the same application reagent was added respectively. After standing at room temperature for 15 min, the absorbance value of each tube was measured at 412 nm wavelength.
Malondialdehyde (MDA) assay kit was required the determination tube (sample group), the opposite tube (50% glacial acetic acid group), blank tube (anhydrous ethanol group), and standard tube (standard substance group) to be set, and the same application reagent was added, respectively. The absorbance value of each tube was measured at 532 nm wavelength after cooling at a 95° water bath for 45 min.
Myeloperoxidase (MPO) assay kit was required measuring tubes (samples) and inverters (distilled water) and adding the same reaction reagent to each tube. Absorbance values of each group are immediately measured at 460 nm wavelength after 10 min of a 60° water bath.
Determination of ROS, CYP2E1, and NADP+/NADPH Ratio
The contents of reactive oxygen species (ROS) and cytochrome P450 2E1 (CYP2E1) in supernatants were detected with enzyme-linked immunosorbent assay (ELISA). The experimental process strictly followed the instructions of the ELISA kit. The fresh liver tissues were homogenized in PBS and centrifuged at 3000 × g at 4 °C for 20 min, retaining the supernatant. 10 μL of distilled water and 10 μL of supernatant was put into the reaction well, added 40 μL diluent and 100 μL Enzyme standard reagent in sequence, mix thoroughly, and incubate at 37° for 60 min, respectively. Then, the liquid in the wells was shaken off, wash solution was added to each well, and the wells were washed 5 times with wash solution. Finally, the stop solution was added, and each group was measured at 450 nm wavelength.
NADP+ is the oxidized form of nicotinamide adenine dinucleotide phosphate (NADPH). NADP+/NADPH is essential for maintaining cellular redox homeostasis and for modulating numerous biological events [17], playing a crucial role in alcohol metabolism [18]. The level of NADPH and NADP+ was evaluated by Coenzyme II(NADP /NADPH) content test kit. Each group was provided two liver tissues of the same volume, dissolved in acidic extract and alkaline extract respectively, thoroughly mixed, centrifuged at 4°10,000 g for 10 min, and the supernatant was retained. Six reagents and supernatant equipped with the kit were added to the control and determined well in sequence. After full mixing, centrifugation was performed at 20,000 g at 25° for 5 min to preserve the precipitation. Then, reagent 7 was added to the precipitate and mixed, and each well was measured with the absorbance value at 570 nm wavelength.
Western Blot Analysis
Total protein from liver tissue was extracted with the protein extraction kits (Beyotime, Jiangsu, Nanjing, China); the nuclear and cytoplasmic proteins were isolated using nuclear extraction kits (Solarbio Science & Technology, Beijing, China). According to the instruction manual, the liver was added to different reagents, homogenized, and centrifuged at 12,000 rpm at 4 °C for 20 min, respectively. And then the concentration of proteins was detected with the Bicinchoninic acid (BCA) protein assay kit (Beyotime, Jiangsu, Nanjing, China). Finally, the protein samples were mixed with the loading buffer and boiled for 15 min. Appropriate protein samples were isolated using 6–10% SDS–polyacrylamide gel electrophoresis (PAGE), transferred onto PVDF membrane and blotted at 4 °C overnight with different primary antibodies: Cleaved caspase 3, Cleaved caspase 8, Cleaved caspase 9, IKKα/β, p-IKKα/β, IкBα, p-IκBα, NF-кB p65, p-NF-кB p65, HO-1 (1:1000, Cell Signaling Technology Inc.), Nrf2 and NQO1(1:1000, Abcam), as well as Bcl-2, Bax, Caspase 3, Caspase 8, Caspase 9, NF-кB p50, Keap1, Lamin B1 and GAPDH (1:1000 Proteintech). The PVDF membranes were incubated with the fluorescence-labeled goat anti-rabbit IgG (1:10,000 Licor) at room temperature under dark light for 1 h. Subsequently, The PVDF was scanned with Odyssey infrared imaging system (Li-cor Bioscience, Lincoln, NE). Total protein and cytoplasmic protein were normalized to GAPDH, and nuclear proteins were normalized to Lamin B1.
RT-PCR Assay
Total RNA was extracted from tissues by RNA Extraction Reagent Kit (Axygen Biotechnology, USA). The preparation and determination of the RT-PCR assay were carried out according to our previous study [15]. The PCR primers were synthesized by Sangon Biotech (Shanghai, China), GAPDH serving as a reference. The primers used in this experiment were listed in Table 1.
Metabolome Profiling
The liver tissue was dissolved in the lysis buffer (formic acid:acetonitrile:water = 10:50:40), and the supernatant was extracted and reserved. The metabolites were separated by ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-Q/TOF MS, Waters, USA). The separation was achieved at 45 °C on an Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 mm; Waters Corporation, Milford, MA, USA). The mass spectra were acquired using a Xevo G2-XS Quadrupole Time-of-Flight mass spectrometer (Waters Corporation, Milford, USA) with Masslynx software v4.1 (Waters Corporation, Milford, USA). The mobile phase was composed of water and acetonitrile (containing 0.1% formic acid). The solvent gradient elution programs of samples were shown in Table 2. A 5 μL volume of sample was injected into the analytical column and the flow rate was 0.4 ml/min.
To ensure the stability and accuracy of the data analysis, 10 μL of supernatant from each sample was mixed as to quality control (QC) samples. Masslynx software v4.1 was used for identifying the original mass spectral data and normalizing the total ion intensity of each chromatogram. The raw data were analyzed by Progenesis QI and EZinfo 3.0 software. The potential metabolites were screened out based on the variable importance (VIP) in the projection values and Student's t-test. VIP > 1 and p < 0.05 were considered as statistically significant. Moreover, the HMDB IDs of metabolites were uploaded to the MetaboAnalyst database (https://www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml) to identify the potential metabolic pathways.
RNA-Seq Profiling
Total RNA was isolated from the livers using Trizol (Thermo Fisher Scientific, USA). The library was constructed with NEBNext UltraTM RNA Library Prep Kit (Illumina) and purified by Library beads (AMPure XP System). NovaSeq 6000 platform was used for sequencing. Reads were processed for quality by Trimmomatic and aligned to the rats’ genome using the STAR alignment algorithm. The differentially expressed genes (DEGs) were identified by the algorithm of DESeq2 FDR analysis; | log2FC |> 1 and P < 0.05 were considered a significant difference.
The volcano plot was drawn by the R language based on the differential gene analysis and the color was determined by the filtering criteria. DEGs were clustered into biological pathways by the DAVID database (https://david.ncifcrf.gov/). Furthermore, DEGs and the HMDB IDs of potential metabolites were submitted to the MetaboAnalyst database (https://www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml) to determine the metabolic pathways.
Statistical Analysis
SPSS software Ver. 24. 0. (Chicago, IL, USA) was performed for statistical analysis. The data for each group were presented as means ± SD and tested by one-way ANOVA with Turkey’s test for post hoc multiple comparisons. In all cases, differences were considered statistically significant with a p-value < 0.05.
Results
AA Treatment Ameliorated Alcohol-Induced Abnormal Liver Function
As shown in Fig. 2a–d, the indication of liver injury was evaluated by serum AST, ALT, TBIL, ALB, and GLB levels. Long-term consumption of alcohol led to a significantly elevated activity of AST, ALT, TBIL, as well as the ratio of ALB/GLB (p < 0.05). However, Silibinin and AA treatment significantly reversed these abnormal changes.

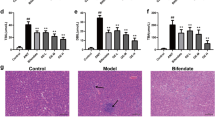
AA treatment ameliorated alcohol-induced abnormal liver function. (a to d) The serum activities of ALT, AST, ALB/GLB, and TBIL were measured using an automatic biochemistry analyzer. (e to h) The contents of TC, TG, HDL, and LDL were detected by commercially available kits. The data are presented as mean ± SD. *P < 0.05 VS. the normal group; #P < 0.05 VS. the model group. (i) H&E staining (200 ×); The red arrow indicated inflammatory infiltration. (k) Masson’s trichrome staining (200 ×)
Furthermore, the contents of TC, TG, and LDL were markedly increased and the content of HDL (p < 0.05) was decreased in the model group. However, Silibinin and AA treatment significantly reversed the expression of these abnormal enzymes (Fig. 2e–h). These results suggested that AA treatment significantly ameliorated alcohol-induced liver function.
AA Attenuated Alcohol-Induced Histomorphological Change
To test the extent of liver tissue damage in rats, H&E staining was examined. As shown in Fig. 2i, liver tissue of the normal control and AA groups showed an intact morphology, clear liver sinuses, without lipid droplets, and inflammatory infiltration. On the contrary, the model group revealed prominent liver damages, as evidenced by the form of slight cytoplasmic microvacuole, loss of cellular boundaries, and even large-scale inflammatory necrosis. However, Silibinin and AA therapy largely alleviated these liver damages.
The result of Masson’s trichrome staining showed that an abundance of collagen deposition was observed in the model group compared to the normal control. While AA and Silibinin treatments significantly reduced collagen accumulation conditions (Fig. 2j). These results demonstrated that AA markedly inhibited the production and accumulation of collagen caused by chronic alcohol.
AA Treatment Restored Metabolic Enzymes Activity
ADH and ALDH are important enzymes for alcohol metabolism. As indicated in Fig. 3a, b, the activity of ADH and ALDH (p < 0.05) was markedly decreased in the model group, while treatment with AA significantly increased the activity of these enzymes.

AA treatment alleviated metabolic enzyme activity and oxidative stress, as well as suppressed pro-inflammatory cytokines. (a and b) The contents of ADH and ALDH were detected by commercially available kits. (c and d) An enzyme-linked immunosorbent assay was used to detect the content of ROS and CYP2E1. (e to g) The contents of NADPH, NADP+ and the ratio of NADP+/NADPH were detected by commercially available kits. (h to l) The contents of MPO, MDA, SOD, GSH-rd, and GSH-px were detected by commercially available kits. (m to o) The mRNA levels of IL-1β, IL6, and TNF-α were detected by the qPCR assay. The data are presented as mean ± SD. *P < 0.05 VS. the normal group; #P < 0.05 VS. the model group
As shown in Fig. 3c–g, chronic alcohol administration caused an increased content of ROS and CYP2E1, as well as the ratio of NADP+/NADPH. However, AA and Silibinin treatment significantly alleviated these phenomena (p < 0.05). These data suggested that AA could alleviate the abnormal expression of metabolic enzymes caused by chronic alcohol administration.
AA Alleviated Alcohol-Induced Hepatic Oxidative Stress
Hepatic oxidative stress was evaluated by measuring myeloperoxidase, lipid peroxidation, and antioxidant function levels. As presented in Fig. 3h, i, chronic alcohol intake led to a significant increase in the myeloperoxidase level of MPO and the lipid peroxidation level of MDA. Meanwhile, the model group had the lowest antioxidant enzyme level of SOD, GSH-rd, and GSH-Px (Fig. 3j–l). However, AA and Silibinin treatment strikingly decreased the contents of MPO and MDA and increased the activities of the antioxidant enzymes including SOD, GSH-Px, and GSH-rd (p < 0.05). These results indicated that AA inhibited chronic alcohol-induced oxidative stress, protecting the liver from damage.
AA Treatment Suppressed Pro-inflammatory Cytokines
Inflammation is one of the main pathogenic factors of alcoholic hepatitis. Compared to the normal, higher mRNA transcription levels of IL-6, IL-1β, and TNF-α (p < 0.05) were observed in the model group. However, the abnormal levels of pro-inflammatory cytokines were significantly alleviated by AA and Silibinin treatment (Fig. 3m–o). These data indicated that AA markedly inhibited pro-inflammatory cytokines release.
AA Treatment Alleviated Alcohol-Induced Hepatocyte Apoptosis
To verify whether alcohol overconsumption affects the process of apoptosis and necrosis, hepatocyte apoptosis was evaluated by TUNEL staining in the present study. As shown in Fig. 4a, there were few TUNEL-positive hepatocytes in the normal group, while numerous TUNEL-positive hepatocytes were observed in the model group. Interestingly, AA and Silibinin treatments significantly reversed these abnormal changes. Similarly, the apoptosis rate of the model group was the highest, while the apoptosis rate of AA and Silibinin groups were observably decreased (p < 0.05) (Fig. 4b).

AA treatment alleviated alcohol-induced hepatocyte apoptosis. (a) TUNEL staining (400 ×); (b) Apoptosis rate of hepatocyte was assessed by TUNEL staining. (c to e) The mRNA levels of BAK, BCL-XL, and MCL-1 were detected by the qPCR assay. (f to i) The protein levels of Bax and Bcl-2 were determined by western blot; The protein levels of Cleaved caspase 3, caspase 3, Cleaved caspase 8, caspase 8, Cleaved caspase 9, and caspase 9 were determined by western blot. The bands 1 to 6 represent the normal control, AA control, Model, Silibinin-treated group, low- and high-dosages of AA treated groups, respectively. The data are presented as mean ± SD. *P < 0.05 VS. the normal group; #P < 0.05 VS. the model group
Compared to the normal group, the transcription level of BAK mRNA was strikingly increased, and BCL-XL and MCL-1 mRNA (p < 0.05) were strikingly decreased in the model group. However, AA and Silibinin treatments significantly reversed these abnormal changes (p < 0.05) (Fig. 4c–e). Additionally, the apoptosis-related factors including the Bcl-2 family and caspases family were detected by Western blotting. As indicated in Fig. 4f–i, an oversupply of alcohol significantly increased the expression of pro-apoptotic protein Bax, cleaved caspase-3, cleaved caspase-8, and cleaved caspase-9, as well as reduced anti-apoptotic protein Bcl-2 expression (p < 0.05). Treatment with AA significantly reversed these abnormal changes.
AA Treatment Activated the Nrf2 Signaling Pathway
Because of the crucial role of the severe oxidative damage in the pathogenesis of alcoholic hepatitis, we further explored the protective mechanism of AH based on the Nrf2-mediated signaling pathway. Nrf2 is a transcription factor that regulates the antioxidant response, and the Nrf2 pathway plays an important role in protecting the liver from damage [19]. As shown in Fig. 5a, b, AA and Silibinin treatments led to a significant up-regulated in the levels of Nrf2, NQO1, and HO-1, while a remarkable decrease in the levels of Keap1 (p < 0.05). Moreover, AA and Silibinin treatments promoted Nrf2 transport from the cytoplasm to the nucleus. Similarly, compared to the model group, the transcription levels of Nrf2, NQO1, HO-1 mRNA were significantly increased, and p62/Sqstm1(p62) was significantly decreased (p 0.05) in AA and Silibinin treatment groups (Fig. 5c–f). Taken together, these data suggested that AA likely activated the Nrf2 signaling pathway, suppressing the oxidative reaction.

AA treatment activated hepatic the Nrf2 signaling pathway. (a) The protein levels of Nrf2, NQO1, HO-1, and Keap 1 were determined by western blot. (b) The cytosol and nucleus protein expressions of Nrf2 were determined by western blot. The bands 1 to 6 represent the normal control, AA control, Model, Silibinin, low- and high-dosages of AA treated groups, respectively. (c to f) The mRNA levels of Nrf2, p62/Sqstm1, NQO1, and HO-1 were detected by the qPCR assay. The data are presented as mean ± SD. *P < 0.05 VS. the normal group; #P < 0.05 VS. the model group
AA Treatment Inhibited Hepatic NF-κB Activation
Alcoholic hepatitis is a severe inflammatory disease, and NF-кB is a key transcriptional factor in regulating the expression of various pro-inflammatory mediators [20]. Therefore, to elucidate the underlying mechanism of AA against inflammatory response, the NF-кB pathway-related key proteins were further detected. As shown in Fig. 6, compared with the normal group, the protein expressions of NF-кB p50, p-NF-кB p65, p-IкBα, and p-IKKα/β were significantly increased (p < 0.05) in the model group. In addition, chronic alcohol abuse promoted NF-кB p65 transport from cytoplasm to the nucleus, and the transcription level of NF-кB p50 and NF-кB p65 mRNA (p < 0.05) was strikingly increased. While AA and Silibinin treatment reversed these abnormal expressions (p < 0.05), indicating that AA inhibited the inflammatory response via inhibiting the NF-кB pathway.

AA treatment inhibited hepatic NF-κB activation. (a) The protein levels of the phosphorylation of NF-κB p65, IKKα/β, and IκBα were determined by western blot. (b) The cytosol and nucleus protein expressions of NF-κB p65 were determined by western blot. (c) The protein level of NF-κB p50 was determined by western blot. The bands 1 to 6 represent the normal control group, AA control, Model, Silibinin, low- and high-dosages of AA treated groups, respectively. (d and e) The mRNA levels of NF-κB p65 and NF-κB p50 were detected by the qPCR assay. The data are presented as mean ± SD. *P < 0.05 VS. the normal group; #P < 0.05 VS. the model group
Transcriptomic Analysis
To explore the effect of AA on the global gene of alcoholic hepatitis in rats, transcriptome analysis was conducted in this study. The Principal Component Analysis (PCA) diagram showed a discrete distribution of genes among the normal, model and AA groups (Fig. 7a). The volcano and heatmap plot indicated that AA enhanced 96 genes expression and inhibited 107 genes expression (Fig. 7b, c). Meanwhile, the KEGG pathway analysis showed that long-term alcoholism might lead to a wide range of metabolic changes in rats because the metabolic pathway was the most relevant one (Fig. 7d). As shown in Fig. 7e, f, AA treatment results in significant transcriptome alterations that may significantly affect metabolic pathways, these data indicated that AA could ameliorate alcohol-induced metabolic disorders at a genetic level.

Transcriptomics analysis between the AA and model group, and integrated analysis of metabolomics and transcriptome. (a) PCA plot; (b) Heatmap; (c) Volcano plot; (d) KEGG pathway analysis for Normal VS model group; (e) KEGG pathway analysis for AA VS model group; (f) Network was built based on the relationship of KEGG Pathways
Metabolome Analysis
As presented in Fig. 8a, the PCA plot of QC samples showed that the points were tightly clustered together, and a similar trend was found in the analysis of Hotelling’s T2 Range (Fig. 8b). These data indicated that the metabolomic analysis method with good repeatability and stability was suitable for the subsequent research.

Metabolome analysis of the normal, AA, and model group. (a) Principal Component Analysis (PCA) diagram of QC; (b) Hotelling’s T2 Range plot; (c) PCA diagram; (d) Volcano plot; (e) Variable importance plot (VIP) for normal VS model group; (f) Metabolic pathways enrichment for the normal and model group (each point represents a metabolic pathway; the size of dot means the impact; the color (red) represents the positive correlation). (g) Variable importance plot (VIP) for AA VS model group; (h) Metabolic pathways enrichment for the AA and model group (each point represents a metabolic pathway; the size of dot means the impact; the color (red) represents the positive correlation); (i) Glycerophospholipid metabolism pathway. Metabolites in red (C00350, C00157, C04230, and C02737) were the potential compounds regulated by the AA
As shown in Fig. 8c, each group formed a distinct cluster in primary component analysis (PCA). And the Volcano plot showed that a total of 1079 metabolites were differentially prominent (p < 0.05) expressed (Fig. 8d). The VIP diagram showed that there were 757 differential metabolites (p < 0.05) between the normal and the model group (Fig. 8e). The MetaboAnalyst analysis revealed that these differential metabolites were mainly enriched in the glycerophospholipid metabolism pathway (P < 0.05 and Impact > 0.4) (Fig. 8f), suggesting that the alcohol-induced AH model was likely to be associated with lipid metabolic disturbance.
Furthermore, 480 differential metabolites were identified between the AA and model groups, of which 16 metabolic pathways were obtained (Fig. 8g, h). Interestingly, the glycerophospholipid metabolism pathway was the most relevant one (P < 0.05 and Impact > 0.2). Further analysis revealed that AA regulated the glycerophospholipid metabolism pathway likely by modulating the synthesis of C00157 (208 PC compounds), C00350 (79 PE compounds), C04230 (5 LysoPC compounds), and C02737 PS(18:0/18:1(9Z)), ultimately ameliorating lipid metabolic disturbance (Fig. 8i).
Integrated Analysis of Transcriptomics and Metabolomics
To explore the systemic mechanism of AA on alcoholic hepatitis, an interaction network between DEGs and differential metabolites was analyzed by the MetaboAnalyst database. As shown in Fig. 9a and Table 3, multiple lipid-related pathways were obtained, such as Retinol metabolism, Glycerophospholipid metabolism, Taurine and hypotaurine metabolism, Linoleic acid metabolism, Steroid hormone biosynthesis (P < 0.05), and Biosynthesis of unsaturated fatty acids (Impact > 0.8). These metabolic pathways may be the key metabolic pathway of AA in treating alcoholic hepatitis. Moreover, glycerophospholipid metabolism was the most relevant metabolic pathway that simultaneously included both the DEGs and differential metabolites. The further analysis revealed that the Gpat3 gene (p < 0.05) might affect the synthesis of four metabolites: LysoPC (1-acyl-Sn-glycerol-3-phosphocholine), PC (Phosphatidylcholine), PE (Phosphatidylethanolamine), and PS (Phosphatidylserine) (Fig. 9b). Besides, compared to the model group, AA greatly decreased the expression of Gpat3 and LysoPC and increased the expression of PC, PS, and PE (Fig. 9c–g). These results suggested that AA might alleviate AH symptoms by regulating lipid metabolism, and Gpat3 may be a key gene in the alteration process.

Integrated analysis of metabolomics and transcriptome. (a) Metabolic pathways enrichment (each point represents a metabolic pathway; the size of the dot means the impact; the color (red) represents the positive correlation). (b) Glycerophospholipid metabolism pathway. Metabolites in red (C00350, C00157, C02737, and C04230) and the GPAT3 gene in yellow were the potential targets regulated by the AA; (c) The quantitative analysis for the DEGs of GPAT3 and metabolites (LysoPC, PC, PE, and PS). The data are presented as mean ± SD. *P < 0.05 VS. the normal group; #P < 0.05 VS. the model group
Discussion
Alcoholic hepatitis is a disease characterized by hepatocyte de-differentiation and loss of hepatocyte regenerative capacity [21]. In our previous studies, we have made a great effort to screen out the potential components of Potentilla chinensis and found that AA may be a candidate drug for the treatment of alcoholic hepatitis. In this study, we tried to elucidate the underlying mechanism of AA against alcoholic hepatitis. The animal model of alcoholic hepatitis was induced by various concentrations of alcohol. We found that AA treatment could significantly attenuate liver damage, as evidenced by the H&E staining with fewer inflammatory necrosis, and the lower activity of ALT, AST, TBIL, and ALB/GLB. Furthermore, AA treatment could significantly reverse the abnormal expression of TC, TG, LDL, and HDL enzyme activities caused by alcohol. Also, Masson’s trichrome staining showed that AA treatment could extremely decrease collagen accumulation. These data indicated that AA greatly inhibited alcohol-induced liver injury.
Considering the important role of apoptosis on alcohol-induced liver injury, we decided to further observe hepatocyte apoptosis by TUNEL staining. On the other hand, the Bcl-2 family of proteins participates in the intrinsic pathway of apoptosis, consisting of anti-apoptotic and pro-apoptotic proteins [22]. The caspase family is the main mediator that activates cell apoptosis; Caspase-3, caspase-8, and caspase-9 are key nodes to control apoptosis in the regulatory network [23]. The present study showed that the model group presented a large number of cell apoptosis, while AA treatment significantly alleviated the condition. Besides, AA markedly inhibited the expression of genes related to the caspase family (Caspase-3, caspase-8, and caspase-9) and pro-apoptotic (Bax and BAK), as well as markedly promoted the expression of genes related to anti-apoptotic (Bcl-2, BCL-XL, and MCL-1). Altogether, these data suggest that AA alleviated hepatocyte apoptosis by regulating the expression of apoptosis-related proteins.
To understand the potential mechanism of AA on hepatitis, alcohol metabolism and inflammatory damage were first considered. Ethanol oxidation mainly regulated by alcohol dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH) leads to excessive ROS and oxidative stress, being the main contributors to alcoholic liver injury [7, 24, 25]. Besides, CYP2E1 is a subfamily of cytochrome P450, and its induction is related to ROS production [26]. Excessive ROS not only further leads to the imbalance of pro-oxidants and antioxidants, but also damages the lipid membrane, which eventually contributes to necrosis or apoptosis [27, 28]. Moreover, the proportion of ethanol is alternatively metabolized by the microsomal ethanol oxidizing system (MEOS) located within the smooth endoplasmic reticulum of hepatocytes, which consumes NADPH, generates NADP+, and dissipates heat to convert into acetaldehyde [29]. In the current study, we found that the contents of ADH and ALDH were considerably enhanced, whereas the activity of ROS and the ratio of NADP+/NADPH were significantly decreased after AA treatment; moreover, AA treatment significantly enhanced the activities of GSH-Rd, GSH-Px, and SOD, while decreased the levels of CYP2E1, MDA, and MPO. These results suggested that AA ameliorated alcohol toxicity by promoting alcohol metabolism, reducing oxidative stress, and restoring the anti-oxidative enzymes.
Generally, inflammation and oxidative stress were crucial inducements to cause alcoholic hepatic injury. In the present study, AA significantly inhibited the expression of the pro-inflammatory cytokines of IL-1β, IL-6, and TNF-α, indicating that AA could effectively reduce the inflammatory infiltration caused by alcohol. Next, we examined the effects of the NF-κB pathway on alcoholic hepatitis model rats. The NF-κB transcription factor is a cell survival regulator, that participated in cancer development by promoting the major inflammatory pathway activated during liver injury [30]. NF-κB is a protein family, composed of RelA (p65), RelB, c-Rel, p50, and p52. In the resting state, NF-κBp65/p50 binds to IκBα to form a polymeric complex, which prevents them from entering the nucleus. The activation of IKKα/β can promote the phosphorylation and degradation of IκBα, which activates NF-κB and separates the polymer formed, releasing p65/p50 heterodimer to regulate the transcription of various genes [31]. In this study, the Western blot analysis showed that AA decreased the expression of NF-κB p50 and the phosphorylation of IκB-α, IKK-α/β, NF-κB p65. Besides, AA also promoted the transfer of NF-κB from the cytoplasm to the nucleus, suggesting that AA may inhibit the NF-κB signaling pathway expression, thus exerting an anti-inflammatory effect.
In addition to the NF-κB signaling pathway, we also focused on the underlying mechanism of AA on oxidative stress, and the Nrf2 signaling pathway was investigated. The p62-Keap1-Nrf2 pathway is a major mechanism in the regulation of cellular redox homeostasis. P62 is a stress-induced cellular protein that possesses several regions that mediate its interactions with other proteins to regulate cellular mechanisms [32]. Nrf2 is a transcription factor that regulates the expression of a series of antioxidant genes to maintain redox homeostasis in the liver, and Keap1 is a key regulator of antioxidant reaction [33]. Under normal physiological conditions, Keap1 stably binds to Nrf2 and immobilizes it in the cytoplasm [19]. When stimulated by inflammatory response or oxidative stress, Nrf2 separates from Keap1 and releases into the nucleus, where p62 interacts with Keap1 [34,35,36]. The present study showed that the transcription levels of Nrf2 and downstream genes NQO1 and HO-1 mRNA significantly were increased after AA treatment, while the expression levels of p62 and Keap1 were significantly decreased. Similarly, AA significantly increased the expression of Nrf2 in the nucleus and significantly decreased the expression of Nrf2 (cytoplasm). The results suggest that AA could alleviate oxidative stress by activating the p62-Keap1-Nrf2 signaling pathway, to some extent.
Furthermore, to further understand the possible mechanisms of AA on alcoholic hepatitis as a whole, transcriptomics and metabolomics were performed in this study. The transcriptomic analysis revealed that there were 207 DEGs between the AA and model groups. Interestingly, the KEGG pathway analysis indicated that these genes were mainly involved in metabolic processes and several metabolic pathways.
According to the results of the transcriptomic analysis, UPLC-Q/TOF–MS was further used to analyze the effect of AA on liver metabolites. We found that AA alleviated liver damage, probably by regulating the glycerophospholipid metabolism pathway. To our knowledge, lipids are the essential metabolites, alterations in hepatic lipid homeostasis may account for the development of hepatic steatosis, which may progress to steatohepatitis, fibrosis, and cirrhosis [37]. There were reports that excess consumption of lysoPCs regulates a variety of biological processes, including cell proliferation and inflammation. Peroxisomes are dynamic organelles surrounded by a single membrane with a defined lipid composition, the major components of which are phosphatidylcholine (PC) and phosphatidylethanolamine (PE) [38]. Besides, PC and PE are important supporting nutrients for the liver, exert an obvious protective effect against liver damage from viruses, pharmaceuticals, alcoholism, and other toxic influences [37, 39]. Phosphatidylserine (PS) is a glycerol phospholipid with negatively charged head groups [40]. In this study, the analysis revealed 480 differential metabolites between the AA and model groups, which were largely enriched in the lipid-related metabolic pathways, and glycerophospholipid metabolism was the most relevant pathway. Interestingly, through a comprehensive analysis of transcriptome and metabolomics, we found that AA significantly inhibited the expression of the Gpat3 gene, which may be the main molecular mechanism of AA's effect on alcoholic hepatitis disease. The expression of the Gpat3 gene was suppressed, thereby affecting the synthesis of the metabolites such as LysoPC (1-acyl-Sn-glycerol-3-phosphocholine), PC (Phosphatidylcholine), PE (Phosphatidylethanolamine), and PS (Phosphatidylserine), and ultimately regulating the glycerophospholipid metabolism pathway. To sum up, AA significantly regulated gene profile and metabolic profile, protecting the liver from damage; the DEGs-mediated glycerophospholipid metabolism pathway might play an important role in the process.
Conclusion
AA significantly ameliorates alcohol-induced liver injury and inflammation, which is likely associated with several aspects (as shown in Fig. 10): (1) activating the Nrf2 signaling pathway to ameliorate oxidative stress; (2) inhibiting the NF-кB pathway to relieve inflammation; (3) attenuating hepatocyte damage by regulating the DEGs-mediated glycerophospholipid metabolism pathway. Our findings contribute to a comprehensive insight into the therapeutical effects of AA on liver damage, which might provide a potential remedy for treating alcoholic hepatitis in the future.

AA activated the Nrf2 and inhibited NF-κB signaling pathway, and promoted Glycerophospholipid metabolism, ultimately alleviating hepatocyte inflammation, apoptosis, and damage in rats with AH
Availability of data and materials
The raw data and materials about this article are available from the corresponding author.
References
Wan, Y., H. Wu, Y. Li, Z. Xu, J. Yang, C. Liu, Y. He, M. Wang, X. Wu, and Y. Zhang. 2020. viaTSG-6 Inhibits Oxidative Stress and Induces M2 Polarization of Hepatic Macrophages in Mice With Alcoholic Hepatitis Suppression of STAT3 Activation. Frontiers in pharmacology 11: 10.
Xiang, X., S. Hwang, D. Feng, V. Shah, and B. Gao. 2020. Interleukin-22 in alcoholic hepatitis and beyond. Hepatology international 14: 667–676.
Takeuchi, M., P. Vidigal, M. Guerra, M. Hundt, M. Robert, M. Olave-Martinez, S. Aoki, T. Khamphaya, R. Kersten, E. Kruglov, et al. 2021. Neutrophils interact with cholangiocytes to cause cholestatic changes in alcoholic hepatitis. Gut 70: 342–356.
Gala, K., and V. Vatsalya. 2020. Emerging noninvasive biomarkers, and medical management strategies for alcoholic hepatitis: present understanding and scope. Cells 9.
Waleed, M., M. Abdallah, Y. Kuo, J. Arab, R. Wong, and A. Singal. 2020. Higher Frequency of Hospital-Acquired Infections but Similar In-Hospital Mortality Among Admissions With Alcoholic Hepatitis at Academic vs. Non-academic Centers. Frontiers in physiology 11: 594138.
Bieghs, V., and C. Trautwein. 2013. The innate immune response during liver inflammation and metabolic disease. Trends in immunology 34: 446–452.
Han, Y., B. Glueck, D. Shapiro, A. Miller, S. Roychowdhury, and G. Cresci. 2020. Dietary Synbiotic Supplementation Protects Barrier Integrity of Hepatocytes and Liver Sinusoidal Endothelium in a Mouse Model of Chronic-Binge Ethanol Exposure. Nutrients 12.
Li, Y., Y. Zeng, Q. Huang, S. Wen, Y. Wei, Y. Chen, X. Zhang, F. Bai, Z. Lu, J. Wei, X. Lin. 2019. Helenalin from Centipeda minima ameliorates acute hepatic injury by protecting mitochondria function, activating Nrf2 pathway and inhibiting NF-κB activation. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 119: 109435.
Zheng, H., J. Wu, H. Huang, C. Meng, W. Li, T. Wei, and Z. Su. 2019. Metabolomics analysis of the protective effect of rubusoside on palmitic acid-induced lipotoxicity in INS-1 cells using UPLC-Q/TOF MS. Molecular omics 15: 222–232.
Rachakonda, V., C. Gabbert, A. Raina, L. Bell, S. Cooper, S. Malik, J. Behari. 2014. Serum metabolomic profiling in acute alcoholic hepatitis identifies multiple dysregulated pathways. PloS one 9: e113860.
Michelena, J., C. Alonso, I. Martínez-Arranz, J. Altamirano, R. Mayo, P. Sancho-Bru, R. Bataller, P. Ginès, A. Castro, and J. Caballería. 2019. Metabolomics Discloses a New Non-invasive Method for the Diagnosis and Prognosis of Patients with Alcoholic Hepatitis. Annals of hepatology 18: 144–154.
Wang, D., L. Lao, X. Pang, Q. Qiao, L. Pang, Z. Feng, F. Bai, X. Sun, X. Lin, and J. Wei. 2018. Asiatic acid from Potentilla chinensis alleviates non-alcoholic fatty liver by regulating endoplasmic reticulum stress and lipid metabolism. International Immunopharmacology 65: 256–267.
Federico, A., M. Dallio, and C. Loguercio. 2017. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules (Basel, Switzerland) 22.
Wei, J., Q. Huang, R. Huang, Y. Chen, S. Lv, L. Wei, C. Liang, S. Liang, L. Zhuo, and X. Lin. 2013. Asiatic acid from Potentilla chinensis attenuate ethanol-induced hepatic injury via suppression of oxidative stress and Kupffer cell activation. Biological pharmaceutical bulletin 36: 1980–1989.
Feng, Z., L. Pang, S. Chen, X. Pang, Y. Huang, Q. Qiao, Y. Wang, S. Vonglorkham, Q. Huang, X. Lin, and J. Wei. Didymin ameliorates dexamethasone-induced non-alcoholic fatty liver disease by inhibiting TLR4/NF-κB and PI3K/Akt pathways in C57BL/6J mice. International immunopharmacology 88: 107003.
Pang, X., Q. Qiao, S. Vonglorkham, Z. Feng, L. Pang, S. Chen, D. Wang, L. Lao, X. Lin, and J. Wei. 2020. Asiatic acid ameliorates acute hepatic injury by reducing endoplasmic reticulum stress and triggering hepatocyte autophagy. Biomed Pharmacother 129: 110375.
Xiao, W., R.S. Wang, D.E. Handy, and J. Loscalzo. 2018. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxidants & Redox Signaling 28: 251–272.
Piszczatowska, K., D. Przybylska, E. Sikora, and G. Mosieniak. Inhibition of NADPH Oxidases Activity by Diphenyleneiodonium Chloride as a Mechanism of Senescence Induction in Human Cancer Cells. Antioxidants 9.
Zhao, X., L. Gong, C. Wang, M. Liu, N. Hu, X. Dai, C. Peng, and Y. Li. 2021. Quercetin mitigates ethanol-induced hepatic steatosis in zebrafish via P2X7R-mediated PI3K/ Keap1/Nrf2 signaling pathway. J Ethnopharmacol 268: 113569.
Sehrawat, T., M. Liu, and V. Shah. 2020. The knowns and unknowns of treatment for alcoholic hepatitis. The lancet Gastroenterology hepatology international 5: 494–506.
Massey, V., A. Parrish, J. Argemi, M. Moreno, A. Mello, M. García-Rocha, J. Altamirano, G. Odena, L. Dubuquoy, A. Louvet, et al. 2021. Integrated Multiomics Reveals Glucose Use Reprogramming and Identifies a Novel Hexokinase in Alcoholic Hepatitis. Gastroenterology 160: 1725-1740.e1722.
Ginting, C., I. Lister, E. Girsang, W. Widowati, D. Yusepany, A. Azizah, and H. Kusuma. 2021. Piper crocatumHepatotoxicity prevention in Acetaminophen-induced HepG2 cells by red betel ( Ruiz and Pav) extract from Indonesia via antioxidant, anti-inflammatory, and anti-necrotic. Heliyon 7: e05620.
Wei, X., H. Wang, X. Sun, X. Huang, W. Xu, Y. Liang, L. Liu, S. Mo, X. Lin, and J. Lin. 2020. 4-hydroxy-2(3H)-benzoxazolone alleviates acetaminophen-induced hepatic injury by inhibiting NF-κB and activating Nrf2/HO-1 signaling pathways. American journal of translational research 12: 2169–2180.
Sung, C.K., S.M. Kim, C.J. Oh, S.A. Yang, B.H. Han, and E.K. Mo. 2012. Taraxerone enhances alcohol oxidation via increases of alcohol dehyderogenase (ADH) and acetaldehyde dehydrogenase (ALDH) activities and gene expressions. Food Chemical Toxicology 50: 2508–2514.
Yoo, Y., E. Jung, H. Kang, I. Choi, K. Choi, and E. Jeung. 2011. The sap of Acer okamotoanum decreases serum alcohol levels after acute ethanol ingestion in rats. International Journal of Molecular Medicine 28: 489–495.
Yu, J., H. Zhu, M. Kindy, and S. Taheri. Cytochrome P450 CYP2E1 Suppression Ameliorates Cerebral Ischemia Reperfusion Injury. Antioxidants 10.
Hsu, M., S. Koike, A. Mello, L. Nagy, and F. Haj. 2020. Hepatic protein-tyrosine phosphatase 1B disruption and pharmacological inhibition attenuate ethanol-induced oxidative stress and ameliorate alcoholic liver disease in mice. Redox biology 36: 101658.
Chuang, C., C. Tsai, E. Lin, C. Huang, Y. Lin, C. Lan, and C. Huang. 2016. Heat-Killed Lactobacillus salivarius and Lactobacillus johnsonii Reduce Liver Injury Induced by Alcohol In Vitro and In Vivo. Molecules 21.
Robinson, K.E., and V.H. Shah. 2020. Pathogenesis and pathways: Nonalcoholic fatty liver disease & alcoholic liver disease. Translational Gastroenterology & Hepatology 5: 49–49.
Shi, X., X. Jiang, B. Yuan, T. Liu, Y. Tang, Y. Che, Y. Shi, and Q. Ai. 2019. LINC01093Upregulation Protects against Alcoholic Hepatitis through Inhibition of NF-κB Signaling Pathway. Molecular Therapy-Nucleic Acids 17: 791–803.
Sun, X., X. Huang, X. Zhu, L. Liu, S. Mo, H. Wang, X. Wei, S. Lu, F. Bai, and D. Wang. et al. HBOA ameliorates CCl-incuded liver fibrosis through inhibiting TGF-β1/Smads, NF-κB and ERK signaling pathways. Biomed Pharmacother 115: 108901.
Li, P., D. Bing, X. Wang, J. Chen, Z. Du, Y. Sun, F. Qi, and H. Chu. New Target of Oxidative Stress Regulation in Cochleae: Alternative Splicing of the p62/Sqstm1 Gene. Journal of molecular neuroscience : MN 2022.
Xu, L., B. Zhao, S. Sun, S. Yu, Y. Wang, R. Ji, Z. Yang, L. Ma, Y. Yao, Y. Chen, et al. 2020. High-dose vitamin C alleviates pancreatic injury via the NRF2/NQO1/HO-1 pathway in a rat model of severe acute pancreatitis. Annals of translational medicine 8: 852.
Wang, K., X. Zheng, Z. Pan, W. Yao, X. Gao, X. Wang, and X. Ding. 2020. Icariin Prevents Extracellular Matrix Accumulation and Ameliorates Experimental Diabetic Kidney Disease by Inhibiting Oxidative Stress via GPER Mediated p62-Dependent Keap1 Degradation and Nrf2 Activation. Frontiers in cell developmental biology 8: 559.
Dkhil, M., A. Abdel Moneim, T. Hafez, M. Mubaraki, W. Mohamed, F. Thagfan, S. and Al-Quraishy. 2019. Myristica fragrans Kernels Prevent Paracetamol-Induced Hepatotoxicity by Inducing Anti-Apoptotic Genes and Nrf2/HO-1 Pathway. Int J Mol Sci 20.
Zhuang, L., W. Ding, Q. Zhang, W. Ding, X. Xu, X. Yu, and D. Xi. 2021. TGR5 Attenuated Liver Ischemia-Reperfusion Injury by Activating the Keap1-Nrf2 Signaling Pathway in Mice. Inflammation 44: 859–872.
Zhou, C., H. Jia, Y. Liu, M. Yu, X. Chang, Y. Ba, and Z. Zou. 2016. Metabolism of glycerophospholipid, bile acid and retinol is correlated with the early outcomes of autoimmune hepatitis. Molecular bioSystems 12: 1574–1585.
Miura, H., H. Mizuguchi, M. Amano-Iwashita, R. Maeda-Kogure, A. Negishi, A. Sakai, T. Toyama, H. Kawai, A. Mitsumoto, and N. Kudo. 2021. Clofibric acid increases molecular species of phosphatidylethanolamine containing arachidonic acid for biogenesis of peroxisomal membranes in peroxisome proliferation in the liver. Biochimica et biophysica acta Molecular and cell biology of lipids 1866: 158963.
Hänninen, S., P. Somerharju, and M. Hermansson. Metabolic Heavy Isotope Labeling to Study Glycerophospholipid Homeostasis of Cultured Cells. Bio-protocol 7: e2268.
Lenoir, G., J. D'Ambrosio, T. Dieudonné, A. Čopič. 2021. Transport Pathways That Contribute to the Cellular Distribution of Phosphatidylserine. Frontiers in cell and developmental biology 9: 737907.
Funding
The authors gratefully acknowledged the financial support provided by the National Natural Science Foundation of China (No. 81660706), the Natural Science Foundation of Guangxi (Nos. 2018GXNSFAA050107), and the Scientific and Technological Innovation Major Base of Guangxi (No. 2018–15-Z04).
Author information
Authors and Affiliations
Contributions
This work was carried out with collaboration among all authors. Jinbin Wei designed the study and was in charge of the funds. Yushen Huang, Hongmei Su, Wuchang Zhu, and Yuanyuan Wei experimented. Yan Long and Yanxia Shi provided help to the experiment. Siyun Chen was responsible for statistical analysis and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments were carried out in strict accordance with the guidelines for the use of animal care in the laboratories of the Experimental Animal Center of Guangxi Medical University. All protocols were approved by an independent animal care and use committee.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, S., Huang, Y., Su, H. et al. The Integrated Analysis of Transcriptomics and Metabolomics Unveils the Therapeutical Effect of Asiatic Acid on Alcoholic Hepatitis in Rats. Inflammation 45, 1780–1799 (2022). https://doi.org/10.1007/s10753-022-01660-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-022-01660-x