
Abstract
The regulatory role of toll-like receptor 4 (TLR4) in the inactivate staphylococcus epidermidis (ISE)-induced cornea inflammation is not well investigated. Here, TLR4 silence could decrease inflammatory cytokines in corneal epithelial cells treated with ISE. The mouse corneal epithelial cells were exposed to ISE for 24 h, either alone or with the NF-κB inhibitor, TLR4 lentivirus to bilaterally (knock-down or and overexpression). The expression of TLR4 in mouse corneal epithelial cells was investigated using western blot and qRT-PCR assay. The inflammatory cytokine levels were evaluated by qRT-PCR and ELISA, respectively. The relative impact factors of TLR4-mediated NF-κB signaling detected using western blot assay. Results show the expression levels of TLR4 and some inflammatory cytokines were significantly increased in corneal epithelial cells treated with ISE. TLR4 Silence markedly decreased ISE-induced production of IL12, TNF-α, CCL5, and CCL9 in corneal epithelial cells. Furthermore, the nuclear translocation of NF-κB p65 and myeloid differentiation protein 88 (MyD88) in the cells treated with ISE were further reduced by silencing TLR4. Inhibition of TLR4-mediated NF-κB signaling by using BAY11-7082 also alleviated ISE-induced inflammation. In the rescue experiment, transfected the stable TLR4 silenced corneal epithelial cells with TLR4 overexpression lentivirus, we found that TLR4 overexpression can restore the down-regulation of TLR4 and inflammatory cytokines (IL12, TNF-α, CCL9) caused by TLR4 knocked down. Therefore, ISE-induced cornea inflammation was due to the activation of the TLR4/MyD88/NF-κB signaling pathway, and dramatically stimulated IL12, TNF-α, CCL9 secretion. TLR4 silence presented mitigates damage in corneal epithelial cells treated with ISE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Infectious keratitis is still a common cause of blindness worldwide [1, 2]. It is estimated that each year has 5 million people in the world suffer from various types of infectious keratitis, and unfortunately, about 20% are blinded due to eye infections [3]. Pathogenic microorganisms which caused microbial keratitis have bacteria, fungi, viruses, or parasites, among them bacterial infection is a common, latently sight threatening cornea inflammation [4, 5]. Comprehensive national conditions, staphylococcus epidermidis accounted for the largest proportion [6, 7]. Thus, we should pay enough attention to it. As a developing country, the incidence of infectious keratitis is higher in China, especially in many rural areas due to poor living conditions and health care awareness [8]. There are the most blind people in China of the world, with about 5 million blind people, accounting for 18% of the world’s blind population, and each year about 45 million people blind in China. Even worse, if the current trend continue to remain the same, the blind people in China is expected 4-fold increase [9]. Therefore, there is an urgent need to prevention of corneal infection. It is a long and arduous task to study the mechanism of immunological regulation and drug development.
The innate immune system recognizes the structural components of pathogens, known as pathogen-associated molecular patterns (PAMP), and the corresponding recognition receptors are called pattern recognition receptors (PRRs) [10,11,12,13]. TLR members are a family of transmembrane receptors that can recognize PAMP, and the subsequent initiation of signal transduction can lead to the release of inflammatory mediators, which plays an important role in natural immune defense [14,15,16,17], and finally activate the adaptive immune system. Therefore, it is believed that TLRs control the transformation from innate immunity to acquired immunity, and it is a pathogen recognition receptor that recently attracted much attention in recent years [18,19,20]. It has been observed that TLRs can interact with MyD88, thereby leading to activate of p38, c-JNK, and NF-κB, which is indispensable for the production of cytokines such as IL-6, IL-8, and TNF-α [21,22,23,24].
TLR4 contains 879 amino acids and is located on the short arm of human chromosome 9. Poltorak et al. first discovered TLR4 in 1998 [25]. TLR4 has multiple functions in the organism and is mainly expressed in CD14+ monocytes and macrophages, while it is also widely expressed in CD14- endothelial cells [26]. TLR4 can recognize exogenous or endogenous pathogen-associated molecular patterns in vivo [27]. TLR4 is an important TLR on the surface of lymphocytes [28]. And it has been a research hotspot in the research fields of oncology, microbiology, and immunology to study the role of TLRs. Studies have shown that TLR4 which expresses in a variety of ocular tissues and cells can identify the particular pathogen and induce inflammatory cytokines expression as well as inflammatory cell infiltration, thereby killing and eliminating pathogens [29]. Gram-negative bacteria transmit infection signals to cells through lipopolysaccharide (LPS), lipopolysaccharide binding protein LBP, lipopolysaccharide receptor CD14, and TLR4, and then mediated by various intracellular signaling pathways, triggering a series of immune responses [30, 31]. However, the regulatory role of TLR4 in corneal infections induced with gram-positive bacteria, such as ISE, remains largely unclear.
Here, we established a corneal epithelial cells model stimulated by ISE, then performed by silencing or and overexpression TLR4 and function of myD88 inhibitors. Herein, we reported that ISE-induced cornea inflammation was due to the activation of the TLR4/MyD88/NF-κB signaling pathway, and subsequently dramatically promoting IL12, TNF-α, CCL9 and CCL5 secretion. These findings revealed a novel immunoregulatory mechanism of TLR4 in the cornea inflammation infected by ISE. This project systematically illustrates the system immunological mechanism of TLR4 in the process of staphylococcus cornea infection. Moreover, this study could provide reference and basic experimental data for the clinical prevention and treatment of infectious keratitis.
METHODS
Reagents
The NF-kB inhibitor, BAY11-7082 was purchased from Merck (Temecula, CA, USA). The anti-rabbit TLR4 polyclonal antibody (ab13556), β-actin (ab8227), and anti-mouse p65 (ab16502), MyD88 (ab2064) were purchased from Abcam (Cambridge, UK). The anti-mouse and anti-rabbit secondary HRP-conjugated antibody for western blot was purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals used in this paper were analytical or reagent grade and were from commercial sources.
Cell Isolation and Culture
The mouse corneal epithelial cells were isolated from corneal tissues of C57BL/6J mice as described previously [32, 33]. In brief, the mice were euthanized by cervical dislocation, then quickly removed eyeballs and washed twice with 1×PBS supplemented with penicillin/streptomycin (Life Technologies, Carlsbad, CA, USA). Next, the corneal tissues were separated on a cold stage under sterile conditions and digested with 0.25% trypsin (Gibco, Carlsbad, CA, USA)/collagenase type IV (0.1%; Sigma, St. Louis, MO, USA) at 37 °C for 30 min. After that, the procured cell suspension was filter with a 100-mesh wire sieve and then centrifuged at 1200 rpm for 3 min. Finally, cells were cultured in DMEM containing with 15% fetal cattle serum (FCS, Hyclone, Logan, UT, USA)); 10 ng/ml epidermal growth factor, IV collagen, and fibronectin; 5 μg/ml recombinant human insulin and human transferrin; 1.8 × 10−10 M adenine, 400 μg/ml hydrocortisone, 10−6 M isoproterenol, 2 × 10−9 M triiodo thyronine, 2 mmol/l glutamine, and 1% penicillin/streptomycin at 37 °C in a 5% CO2 incubator. Cells were cultured and identified and as a subject for further study. For induction of inflammation, corneal epithelial cells were exposed to ISE for 24 hours. To further examine the mechanism of TLR4 in ISE-induced cornea inflammation, corneal epithelial cells were pre-incubated with 10 μM BAY11-7082 for 2 h before ISE treatment.
Construction of Recombined Lentivirus Vector and Transfection
The CDS sequence of TLR4 was downloaded from GenBank (accession no. AF177767.1). In order to silence the expression of TLR4, we obtained three candidate shRNAs and one scrambled shRNA by predicted on the line software (http://biodev.extra.cea.fr/DSIR/DSIR.html), and packaged recombined lentivirus. And at the meantime, the CDS sequence of TLR4 was sub-cloned into the BamH1, XhoI, and Notl sites of pCDH1-CMV-BXN-EF1-Neo vector (Invivogen, San Diego, CA) to generate the pCDH1-TLR4 plasmid (recombinant expression vector of TLR4), and packaged recombined lentivirus. The empty pCDH1-CMV-BXN-EF1-Neo vector was employed as the negative control. The primers applied to construct the TLR4 recombined plasmid are shown in Table 1. Transfection of TLR4 overexpression construct or TLR4 shRNAs lentivirus as well as the negative control after ISE was performed. Transfection was confirmed by qRT-PCR and Western blot analysis.
Total RNA Isolation and Quantitative Real-time PCR (qRT-PCR)
Total cellular RNA was isolated from corneal epithelial cells by using Trizol Reagent (Invitrogen, Carlsbad, CA) under product instruments. Total RNA purity was detected by using an OneDrop 1000 spectrophotometer (Wuyi technology. Nanjing, China) at the 260/280 nm ratio. Then, the RNA (1 μg) was subsequently reverse transcribed into cDNA using a Reverse Transcription Kit (Takara, Kusatsu, Japan). To check the mRNA expression level of TLR4, qRT-PCR was performed triplicate with the SYBR Green Master Mixture (Takara, Kusatsu, Japan) on a Lightcycler 96 system (Roche, Basel, Switzerland). The thermal cycling conditions comprised 5 min at 95 °C, 45 cycles at 95 °C for 20 s, 56 °C for 15 s, and 72 °C for 20 s. The expression of genes was analyzed using the 2−△△CT method and normalized using GAPDH. The Primer sequences used for qRT-PCR are listed in Table 2.
Western Blot Analysis
Total cellular proteins were harvested from corneal epithelial cells stimulated with ISE in the absence or presence of 10 μM BAY11-7082 by using the RIPA lysis buffer. The BCA assay was adopted to determine the concentration of protein. A total of 20 μg protein diluted in 5×SDS loading buffer were boiled for 5 min and subjected to 12% SDS-PAGE gels. And the separated proteins were transferred to PVDF membranes (Bio-Rad, Hercules, CA) followed by blocking with 5% fat-free powdered milk in Tris-buffer saline and 0.1% Tween 20 (TBST) for 2 h at room temperature. Membranes were immunoblotted with primary antibodies against TLR4 (1:500), NF-κB p65 (1:500), MyD88 (1:500), or β-actin (1:500, internal control) overnight at 4 °C. Following washed thrice with TBST, the membranes were incubated with appropriate secondary HRP-conjugated antibodies (1:5000) for 2 h at room temperature. After washing, immunoreactive signals were detected using an ECL Kit, and scanned by chemiluminescence detection system (Tanon, Shanghai, China). Image J (NIH) was performed to analysis the bands. All experiments were conducted at least three times in triplicate.
Cytokine Studies
To examine the secretion of cytokines, corneal epithelial cells were stimulated with ISE in the absence or presence of 10 μM BAY11-7082 for 24 h. The cell-free supernatants were collected by centrifugation at 1000 rpm for 10 min. The secretion of IL1 (cat. No. ml027163, Inc. Biorbyt), IL2 (cat. No. KE10004, Inc. proteintech), IL12 (cat. No. KE10014, Inc. proteintech), INF-α (cat. No.ML1138, Inc. Abnova), TNF-a (cat. No. 29832165. Inc. eBioscience), CXCL1 (cat. No. KE10019. Inc. proteintech), CXCL2 (cat. No. KE10022. Inc. proteintech), CCL5 (cat. No. ml024686. Inc. Biorbyt), and CCL9 (cat. No. EK1225. Inc. Boster) in cell-free supernatants of different groups was evaluated using the commercial corresponding ELISA Reagent kits according the manufacturer’s instructions. Absorbance was measured at 450 nm using ELX-800 microplate reader (BioRad, Hercules, CA).
Statistical Analysis
Values are reported as mean ± standard deviation (SD). Each treatment experiment was performed at least three times in triplicate. Data were analyzed using GraphPad Prism 5. The results were analyzed using One-way ANOVA and t test. P < 0.05 was considered to be statistically significantly.
RESULTS
ISE-induced TLR4 and Related Inflammatory Cytokine Expression in Corneal Epithelial Cells
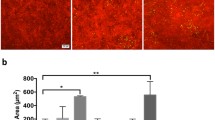
Initially, we evaluated the expression of TLR4 in corneal epithelial cells after exposed to ISE for 24 h, and then collected the total RNA and protein of the cells. The expression levels of TLR4 were detected by qRT-PCR and western blot. As expected, the results indicated that the mRNA and protein levels of TLR4 were upregulated significantly in ISE treatment group compared with the control group (Fig. 1a–c). We next assessed the effects of ISE on the transcription level of inflammatory cytokines. As shown in Fig. 1d, the qRT-PCR assay declared that the inflammatory cytokines gene IL2, IL12, TNF-α, CXCL1, CCL9, and CCL5 levels of in corneal epithelial cells were increased significantly, and many studies have suggested that overstimulation of these cytokines can result in scar formation in the cornea and eventually bring to blindness [34]. However, no changes were discovered in the transcription level of IL1, INF, and CXCL2. The results indicated that corneal epithelial cells infected with ISE can activate the expression of TLR4 and subsequently promote the transcriptional level of multiple inflammatory cytokines.

ISE-induced TLR4 and related inflammatory cytokines expression in corneal epithelial cells. The corneal epithelial cells were exposed to ISE for 24 h, and then collected the total RNA and protein of the cells. a TLR4 mRNA level was measured by qRT-PCR. b TLR4 protein level was determined by western blot. TLR4 was upregulated at both gene and protein levels. c The transcription level of inflammatory cytokines measured by qRT-PCR. Date are presented as mean ± SD (error bars).* p < 0.05, **p < 0.01, and ***p < 0.001 vs Control.
TLR4 Silence Reduced the Secretion of Related Inflammatory Cytokines in Corneal Epithelial Cells
To investigate the role of TLR4 in ISE-induced keratitis, the corneal epithelial cells were transfected with lentivirus to knock-down the expression of TLR4. qRT-PCR and western blot analysis displayed that the transcriptional and protein levels of TLR4 were markedly decreased (Fig. 2a–c). In addition, we analyzed the secretion of related inflammatory cytokines in cell-free supernatants. The ELISA assay demonstrated that TLR4 silence declined the secretion of inflammatory cytokines, especially inflammatory IL12, TNF-α, CCL5, and CCL9 secretion were dramatically reduced (Fig. 2d). Together, the results suggest that ISE induced the inflammatory response in corneal epithelial cells by activating the TLR4.

TLR4 silence reduced the secretion of related inflammatory cytokines in corneal epithelial cells. TLR4 shRNA knock-down vector was constructed, and packaged recombined lentivirus. ELISA kits was used to detect the changes of various inflammatory factors in cells treated with ISE after TLR4 silenced. Three TLR4 shRNA lentivirus were packaged and transfected into corneal epithelial cells. a Extract RNA to detect TLR4 mRNA level by qRT-PCR; TLR4 mRNA levels were significantly down-regulated. b Protein was extracted, and the TLR4 protein level was down-regulated tested by western blot. c Statistical graph of figure b. d After TLR4 silenced, cell-free supernatants were collected for detected the relevant inflammatory factors by ELISA, in which IL12, TNF-α, CCL5, and CCL9 were significantly down-regulated. Date are presented as mean ± SD (error bars).*p < 0.05, **p < 0.01 and ***p < 0.001 vs shControl.
ISE-induced Cornea Inflammation via the TLR4/MyD88/NF-κB Signaling Pathway
We next extended our study to illuminate the mechanisms by which ISE induces the inflammatory response in corneal epithelial cells. NF-κB was a potentially downstream molecule of activated TLR4 which was occurring through MyD88. NF-κB has been reported to be linked to various cell processes, such as pro-inflammation, cell proliferation, angiogenesis, and inhibition of apoptosis [35]. We confused whether NF-κB signaling pathway is involved in inflammatory response induced by ISE. Therefore, corneal epithelial cells were silenced the expression of TLR4 before stimulated with ISE. We noted that NF-κB p56 and MyD88 expression at corneal epithelial cells was lower in TLR4 silence group (shTLR4-1/2) than that in normal control group (Control) (Fig. 3a, c). To further test whether the ISE-induced inflammatory response was result from the activation of NF-κB signaling pathway. As depicted in Fig. 3b, d, the NF-κB p65 and MyD88 protein levels in ISE treatment group (ISE) were significantly increased compared with Control group (p < 0.01). As expected, in the presence of an NF-κB inhibitor BAY11-7082 before treated with ISE dramatically abolished this effect. To further explore the effect of the activation of NF-κB signaling pathway on inflammatory cytokine secretion, we found that the increased IL12, TNF-α, and CCL9 levels in cells and cell-free supernatants stimulated with ISE were reversed in the presence of BAY11-7082 which detected by qPCR and ELISA, respectively. Our observation suggested that the activation of TLR4 triggered the inflammatory response via MyD88/NF-κB signaling pathway in corneal epithelial cells treated with ISE.

ISE-induced cornea inflammation via the TLR4/MyD88/NF-κB signaling pathway. The expression of MyD88 and P65 in TLR4-mediated NF-κB signaling pathway. Mechanism of inflammation in corneal epithelial cells, a Cell proteins of the control group and TLR4 knock-down group (shTLR4-1/2) were collected after ISE treatment, respectively, and NF-κB signal was detected. b Cells were divided into untreated group (control), ISE treated group (ISE) and ISE+ BAY11-7082 (an NF-κB inhibitor, 10 μM) treated group (ISE+ BAY11-7082), then the expression of p65 and MyD88 in each group was detected by western blot. c Statistical graph of figure a. d Statistical graph of figure b. e The cells were divided into control, ISE and ISE+ BAY11-7082 groups, and the expressions of IL12, TNF-α, and CCL9 were detected by qPCR and ELISA, respectively. Date are presented as mean ± SD (error bars).*p < 0.05, **p < 0.01 vs Control, #p < 0.05 vs ISE.
TLR4 Overexpression Restored the Down-regulation of Inflammatory Cytokines Caused by TLR4 Silence
Our previous research revealed ISE triggers the inflammatory response in corneal epithelial cells by activating the TLR4. In order to further solidify the role of TLR4 in cornea inflammation induced by ISE, we construct the pCDH1-TLR4 overexpression plasmid, and packaged recombined lentivirus in the following rescue experiment. TLR4 was markedly upregulated in RNA and protein levels in corneal epidermal cells transfected with TLR4 recombined lentivirus (TLR4 overexpression, TLR4-OE) compared with the negative control group (Fig. 4a–c). As we know from Fig. 2d, TLR4 silence significantly reduced the secretion of IL12, TNF-α, CCL5, and CCL9 in ISE-treated cells. To confirm this effect was caused by TLR4 silence in this cells, TLR4 knock-down was rescued by TLR4-OE lentivirus and then examined the transcriptional levels of gene IL12, TNF-α, and CCL9. As shown in Fig. 4d, the down-regulation of IL12, TNF-α, and CCL9 in TLR4 silence groups (ISE+shTLR4-1/2) were reversed by TLR4 overexpression (ISE+shTLR4-1/2+TLR4-OE), which was verified by qRT-PCR. In other words, TLR4 overexpression restores the down-regulation of inflammatory cytokines caused by TLR4 silence in corneal epidermal cells stimulated with ISE. These outcomes suggest that activation of TLR4 triggered the inflammatory response via MyD88/NF-κB signaling pathway in corneal epithelial cells treated with ISE, and thus dramatically stimulated IL12, TNF-α and CCL9 secretion.

TLR4 overexpression restored the down-regulation of inflammatory cytokines caused by TLR4 knockdown. Rescue experiment of TLR4 overexpression. Constructed the TLR4 overexpression plasmid, packaged lentivirus, and transfection was confirmed by qRT-PCR and Western blot analysis. a TLR4 mRNA level was measured by qRT-PCR. b TLR4 protein level was determined by western blot. c Statistical graph of figure b. TLR4 was upregulated at both RNA and protein levels. d Further verify whether TLR4 overexpression can restore the down-regulation of some inflammatory factors caused by TLR4 knockdown. TLR4 knockdown cells which treated with ISE were transfected with TLR4 overexpression plasmid lentivirus, then expression of related inflammatory factors was verified at mRNA levels. Date are presented as mean ± SD (error bars).*p < 0.05, **p < 0.01, and ***p < 0.001 vs Control.
DISCUSSION
Bacterial keratitis is corneal infection mainly caused by Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus pneumonia, and other pathogen [6], and which is the major cause of blindness in both developing counties and developed countries. The number of blindness and visual morbidity is projected to increase. Nevertheless, the overuse of antibiotics limits the effectiveness of antimicrobial therapy; it is urgently needed to develop additional therapeutic strategies in addition to antimicrobial therapy [7, 36].
TLRs play an important role in recognizing PAMP and are sentinels of innate immunity, which then leads to the development of adaptive immune responses. TLR4, one important member of TLRs, also plays a key role in the inflammatory response mediated by the NF-κB signaling pathways [37, 38]. According to reports, human corneal epithelial cells stimulated with bacterially derived products such as lipopolysaccharides (LPS) can express TLR4 and secrete pro-inflammatory factors [39, 40]. MyD88 took part in inflammatory cytokine production during pathogen infection via the activation of NF-kB. TLR4 and MyD88 are upstream regulators of NF-kB [41]. NF-kB is a nuclear transcription factor which regulates the expression of inflammation-related genes. NF-kB was isolated in an inactive form in the cytoplasm by binding to the IkB complex. After activation, NF-kB translocated to the nucleus, starting gene transcription in the nucleus, and producing various cytokines [42, 43]. In previous studies, gram-negative bacteria can transmit infection signals to cells and then mediated by various intracellular signaling pathways and trigger a series of immune responses in corneal epithelial cells [44]. However, the regulatory role of TLR4 in corneal infections induced with gram-positive bacteria, such as ISE, remains largely unclear.
In the present study, we sought to investigate the effects of TLR4 on the ISE-induced inflammation in corneal epithelial cells through a series of in vitro experiments. The present study showed that TLR4 as well as inflammation-related genes were upregulated dramatically in ISE-induced corneal epithelial cells. And the silence of TLR4 not only suppressed the secretion of inflammatory cytokines but also the protein levels of NF-κB p56 and MyD88 in ISE-induced corneal epithelial cells, in the other word, the underlying mechanism of ISE-induced inflammatory response in corneal epithelial cells may be the activation of TLR4/MyD88/NF-κB signaling pathway. To further explore the mechanism underlying ISE-induce the inflammatory response in corneal epithelial cells, we noted that NF-κB p56 and MyD88 expression was increased in ISE-induced corneal epithelial cells which was abolished by pre-administration of BAY11-7082. Moreover, in the final rescue experiment, TLR4 overexpression restored the down-regulation of inflammatory cytokines caused by TLR4 silence. Thus, the previous results showed that the activation of TLR4 triggered the inflammatory response via MyD88/NF-κB signaling pathway in corneal epithelial cells treated with ISE.
In conclusion, our study proposed a novel mechanism of ISE-induced inflammation in corneal epidermal cells, which was that the activation of TLR4 triggered the inflammation response via activating the MyD88/NF-κB signaling pathway. Moreover, our date may be promising for the therapeutic approach aiming to preventing ISE-induced inflammation of corneal by target silencing TLR4.
References
Bagnall, K., J. Raso, M. Moreau, J. Mahood, X. Wang, and M. Beuerlein. 2002. The development of scoliosis following pinealectomy in young chickens is not the result of an artifact of the surgical procedure. Studies in Health Technology and Informatics 88: 3–9.
Austin, A., T. Lietman, and J. Rose-Nussbaumer. 2017. Update on the management of infectious keratitis. Ophthalmology 124: 1678–1689.
Chen, J., and J. Yuan. 2007. Stress on recognition and standardizing medical therapy for infectious corneal diseases in China. Ophthalmology in China 16 (3): 145–147.
Collier, S.A., M.P. Gronostaj, A.K. MacGurn, J.R. Cope, K.L. Awsumb, J.S. Yoder, M.J. Beach, and Centers for Disease, C., & Prevention. 2014. Estimated burden of keratitis—United States, 2010. MMWR. Morbidity and Mortality Weekly Report 63 (45): 1027–1030.
Berger, E.A. 2019. Understanding the role of pro-resolving lipid mediators in infectious keratitis. Advances in Experimental Medicine and Biology 1161: 3–12.
Lichtinger, A., S.N. Yeung, P. Kim, M.D. Amiran, A. Iovieno, U. Elbaz, J.Y. Ku, R. Wolff, D.S. Rootman, and A.R. Slomovic. 2012. Shifting trends in bacterial keratitis in Toronto: an 11-year review. Ophthalmology 119 (9): 1785–1790.
Chang, V.S., D.K. Dhaliwal, L. Raju, and R.P. Kowalski. 2015. Antibiotic resistance in the treatment of staphylococcus aureus keratitis: a 20-year review. Cornea 34 (6): 698–703.
Khor, W.B., V.N. Prajna, P. Garg, J.S. Mehta, L. Xie, Z. Liu, M.D.B. Padilla, C.K. Joo, Y. Inoue, P. Goseyarakwong, F.R. Hu, K. Nishida, S. Kinoshita, V. Puangsricharern, A.L. Tan, R. Beuerman, A. Young, N. Sharma, B. Haaland, F.S. Mah, E.Y. Tu, F.J. Stapleton, R.L. Abbott, D.T. Tan, and Group, A. 2018. The Asia Cornea Society Infectious Keratitis Study: a prospective multicenter study of infectious keratitis in Asia. American Journal of Ophthalmology 195: 161–170.
Gerke, J.R., and M.V. Magliocco. 1971. Experimental Pseudomonas aeruginosa Infection of the Mouse Cornea. Infection and Immunity 3 (2): 209–216.
Kufer, T.A., E.M. Creagh, and C.E. Bryant. 2019. Guardians of the Cell: Effector-triggered immunity steers mammalian immune defense. Trends in Immunology 40 (10): 939–951.
Wang, C., G. Wang, C. Zhang, P. Zhu, H. Dai, N. Yu, Z. He, L. Xu, and E. Wang. 2017. OsCERK1-mediated chitin perception and immune signaling requires receptor-like cytoplasmic kinase 185 to activate an MAPK Cascade in Rice. Molecular Plant 10 (4): 619–633.
Segonzac, C., A.P. Macho, M. Sanmartin, V. Ntoukakis, J.J. Sanchez-Serrano, and C. Zipfel. 2014. Negative control of BAK1 by protein phosphatase 2A during plant innate immunity. The EMBO Journal 33 (18): 2069–2079.
Kadota, Y., J. Sklenar, P. Derbyshire, L. Stransfeld, S. Asai, V. Ntoukakis, J.D. Jones, K. Shirasu, F. Menke, A. Jones, and C. Zipfel. 2014. Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Molecular Cell 54 (1): 43–55.
Goodwin, M., E. Lee, U. Lakshmanan, S. Shipp, L. Froessl, F. Barzaghi, L. Passerini, M. Narula, A. Sheikali, C.M. Lee, G. Bao, C.S. Bauer, H.K. Miller, M. Garcia-Lloret, M.J. Butte, A. Bertaina, A. Shah, M. Pavel-Dinu, A. Hendel, M. Porteus, M.G. Roncarolo, and R. Bacchetta. 2020. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Science Advances 6 (19): eaaz0571.
Franz, K.M., and J.C. Kagan. 2017. Innate immune receptors as competitive determinants of cell fate. Molecular Cell 66 (6): 750–760.
Rodet, F., A. Tasiemski, C. Boidin-Wichlacz, C. Van Camp, C. Vuillaume, C. Slomianny, and M. Salzet. 2015. Hm-MyD88 and Hm-SARM: two key regulators of the neuroimmune system and neural repair in the medicinal leech. Scientific Reports 5: 9624.
Kargas, V., J.K. Marzinek, D.A. Holdbrook, H. Yin, R.C. Ford, and P.J. Bond. 2017. A polar SxxS motif drives assembly of the transmembrane domains of toll-like receptor 4. Biochimica et Biophysica Acta - Biomembranes 1859 (10): 2086–2095.
Su, L.C., W.D. Xu, and A.F. Huang. 2020. IRAK family in inflammatory autoimmune diseases. Autoimmunity Reviews 19: 102461.
Leventhal, J.S. 2018. Lose appetite, lose control: integrins and noncanonical autophagy regulate germinal center reactions. The Journal of Clinical Investigation 128 (9): 3752–3753.
Dinarello, C.A. 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunological Reviews 281 (1): 8–27.
Rangasamy, S.B., M. Jana, A. Roy, G.T. Corbett, M. Kundu, S. Chandra, S. Mondal, S. Dasarathi, E.J. Mufson, R.K. Mishra, C.H. Luan, D.A. Bennett, and K. Pahan. 2018. Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer's pathology. The Journal of Clinical Investigation 128 (10): 4297–4312.
Sintes, J., M. Gentile, S. Zhang, Y. Garcia-Carmona, G. Magri, L. Cassis, D. Segura-Garzon, A. Ciociola, E.K. Grasset, S. Bascones, L. Comerma, M. Pybus, D. Llige, I. Puga, C. Gutzeit, B. He, W. DuBois, M. Crespo, J. Pascual, A. Mensa, J.I. Arostegui, M. Juan, J. Yague, S. Serrano, J. Lloreta, E. Meffre, M. Hahne, C. Cunningham-Rundles, B.A. Mock, and A. Cerutti. 2017. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nature Communications 8 (1): 1462.
Schweighoffer, E., J. Nys, L. Vanes, N. Smithers, and V.L.J. Tybulewicz. 2017. TLR4 signals in B lymphocytes are transduced via the B cell antigen receptor and SYK. The Journal of Experimental Medicine 214 (5): 1269–1280.
Ignatz-Hoover, J.J., H. Wang, S.A. Moreton, A. Chakrabarti, M.K. Agarwal, K. Sun, K. Gupta, and D.N. Wald. 2015. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia 29 (4): 918–926.
Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282 (5396): 2085–2088.
Yang, W.S., J.J. Kim, M.J. Lee, E.K. Lee, and S.K. Park. 2018. ADAM17-mediated Ectodomain shedding of toll-like receptor 4 as a negative feedback regulation in lipopolysaccharide-activated aortic endothelial cells. Cellular Physiology and Biochemistry 45 (5): 1851–1862.
Freise, N., A. Burghard, T. Ortkras, N. Daber, A. Imam Chasan, S.L. Jauch, O. Fehler, J. Hillebrand, M. Schakaki, J. Rojas, B. Grimbacher, T. Vogl, A. Hoffmeier, S. Martens, J. Roth, and J. Austermann. 2019. Signaling mechanisms inducing hyporesponsiveness of phagocytes during systemic inflammation. Blood 134 (2): 134–146.
Bourke, E., D. Bosisio, J. Golay, N. Polentarutti, and A. Mantovani. 2003. The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood 102 (3): 956–963.
Wada, J., and H. Makino. 2016. Innate immunity in diabetes and diabetic nephropathy. Nature Reviews. Nephrology 12 (1): 13–26.
Skirecki, T., and J.M. Cavaillon. 2019. Inner sensors of endotoxin - implications for sepsis research and therapy. FEMS Microbiology Reviews 43 (3): 239–256.
He, Y., S. Liu, D.E. Kling, S. Leone, N.T. Lawlor, Y. Huang, S.B. Feinberg, D.R. Hill, and D.S. Newburg. 2016. The human milk oligosaccharide 2’-fucosyllactose modulates CD14 expression in human enterocytes, thereby attenuating LPS-induced inflammation. Gut 65 (1): 33–46.
Zhang, W., W. Li, C. Zhang, C. Zhu, X. Yi, Y. Zhou, and Y. Lv. 2019. Effects of vitamin A on expressions of apoptosis genes Bax and Bcl-2 in epithelial cells of corneal tissues induced by benzalkonium chloride in mice with dry eye. Medical Science Monitor 25: 4583–4589.
Chawla, S., and S. Ghosh. 2018. Establishment of in vitro model of corneal scar pathophysiology. Journal of Cellular Physiology 233 (5): 3817–3830.
Huxlin, K.R., H.B. Hindman, K.I. Jeon, J. Buhren, S. MacRae, M. DeMagistris, D. Ciufo, P.J. Sime, and R.P. Phipps. 2013. Topical rosiglitazone is an effective anti-scarring agent in the cornea. PLoS One 8 (8): e70785.
Li, Y., J.Y. Yang, X. Xie, Z. Jie, L. Zhang, J. Shi, D. Lin, M. Gu, X. Zhou, H.S. Li, S.S. Watowich, A. Jain, S. Yun Jung, J. Qin, X. Cheng, and S.C. Sun. 2019. Preventing abnormal NF-kappaB activation and autoimmunity by Otub1-mediated p100 stabilization. Cell Research 29 (6): 474–485.
Lakhundi, S., R. Siddiqui, and N.A. Khan. 2017. Pathogenesis of microbial keratitis. Microbial Pathogenesis 104: 97–109.
Wang, W., Z. Deng, H. Wu, Q. Zhao, T. Li, W. Zhu, X. Wang, L. Tang, C. Wang, S.Z. Cui, H. Xiao, and J. Chen. 2019. A small secreted protein triggers a TLR2/4-dependent inflammatory response during invasive Candida albicans infection. Nature Communications 10 (1): 1015.
Brennan, J.J., and T.D. Gilmore. 2018. Evolutionary Origins of Toll-like Receptor Signaling. Molecular Biology and Evolution 35 (7): 1576–1587.
Engelmann, C., M. Sheikh, S. Sharma, T. Kondo, H. Loeffler-Wirth, Y.B. Zheng, S. Novelli, A. Hall, A.J.C. Kerbert, J. Macnaughtan, R. Mookerjee, A. Habtesion, N. Davies, T. Ali, S. Gupta, F. Andreola, and R. Jalan. 2020. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. Journal of Hepatology 73: 102–112.
Kang, K., M. Bachu, S.H. Park, K. Kang, S. Bae, K.H. Park-Min, and L.B. Ivashkiv. 2019. IFN-gamma selectively suppresses a subset of TLR4-activated genes and enhancers to potentiate macrophage activation. Nature Communications 10 (1): 3320.
Zhou, Y., J. Ming, M. Deng, Y. Li, B. Li, J. Li, Y. Ma, Z. Chen, and S. Liu. 2020. Berberine-mediated up-regulation of surfactant protein D facilitates cartilage repair by modulating immune responses via the inhibition of TLR4/NF-kB signaling. Pharmacological Research 155: 104690.
Limongi, D., S. Baldelli, P. Checconi, M.E. Marcocci, G. De Chiara, A. Fraternale, M. Magnani, M.R. Ciriolo, and A.T. Palamara. 2019. GSH-C4 acts as anti-inflammatory drug in different models of canonical and cell autonomous inflammation through NFkappaB Inhibition. Frontiers in Immunology 10: 155.
Kwon, J.W., H.K. Kwon, H.J. Shin, Y.M. Choi, M.A. Anwar, and S. Choi. 2015. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-kappaB. Scientific Reports 5: 14470.
Brothers, K.M., J.D. Callaghan, N.A. Stella, J.M. Bachinsky, M. AlHigaylan, K.L. Lehner, J.M. Franks, K.L. Lathrop, E. Collins, D.M. Schmitt, J. Horzempa, and R.M.Q. Shanks. 2019. Blowing epithelial cell bubbles with GumB: ShlA-family pore-forming toxins induce blebbing and rapid cellular death in corneal epithelial cells. PLoS Pathogens 15 (6): e1007825.
Funding
This study was supported by grants from the Natural Science Foundation of Jiangsu Province, China (15KJB180015), Nantong Science and Technology Program (JC2018090), and Scientific Innovation Research of College Graduates in Jiangsu Province (KYCX18-2415).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
We hereby declare that there is no conflict of interest between the authors of this manuscript.
Ethical Approval
All animal procedures were approved by the Lab Animal Ethical Committee of Nantong University and were performed according to the Standard Operating Procedures for Laboratory Animal Center of Nantong University.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, L., Du, L., Ju, Q. et al. Silencing TLR4/MyD88/NF-κB Signaling Pathway Alleviated Inflammation of Corneal Epithelial Cells Infected by ISE. Inflammation 44, 633–644 (2021). https://doi.org/10.1007/s10753-020-01363-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-020-01363-1