
Abstract
MCC950 has been proposed as a specific small molecule inhibitor that can selectively block NLRP3 inflammasome activation. However, the exact mechanism of its action is still ambiguous. Accumulating investigations imply that chloride efflux–dependent ASC speck oligomerization and potassium efflux–dependent activation of caspase-1 are the two relatively independent, but indispensable events for NLRP3 inflammasome activation. Previous studies suggested that influence of MCC950 on potassium efflux and its consequent events such as interaction between NEK7 and NLRP3 are limited. However, inhibiting chloride intracellular channel–dependent chloride efflux leads to a modification of inflammatory response, which is similar to the function of MCC950. Based on these findings, we shed new insights on the understanding of MCC950 that its function might correlate with chloride efflux, chloride intracellular channels, or other targets that act upstream of chloride efflux.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
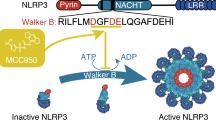
Inflammasomes are a group of multi-protein complex that play key roles in inflammation and immunity. In most cases, PRR (pattern recognition receptor) as the sensor, the adaptor ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)), and cysteine protease caspase-1 are the three essential components for inflammasome assembly [1]. Three members of NLR (NOD-like receptor) family (NLRP1, NLRP3, and NLRC4) and AIM2 (absent in melanoma 2) are all important PRRs that can form the inflammasomes [2, 3]. NLRP3 has a tripartite structure including the LRRs (leucine-rich repeats) that are involved in recognition of stimuli; NACHT (nucleotide-binding-and-oligomerization domain) that mediates self-oligomerization; and PYD (pyrin domain) that mediates the interaction with ASC (Fig. 1) [1]. Apart from these three domains, NLRP1 and NLRC4 (lacks the PYD) also contain a CARD, so they can interact with pro-caspase in the absence of ASC. AIM2 has a PYD that mediates the interaction with ASC and a HIN-200 domain that recognizes dsDNA [4].

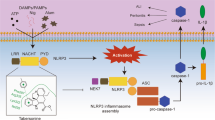
Schematic diagram of NLRP3 inflammasome assembly. Left part: canonical inflammasome. Inflammatory stimuli such as PAMPs and DAMPs are recognized by TLRs, which in turn induce NF-κB-mediated upregulation of NLRP3, pro-caspase-1, pro-IL-1β, and pro-IL-18 (priming step). Potassium efflux leads to forming a complex by NEK7 with NLRP3 (NLRP3–NEK7). Chloride efflux causes oligomerization of ASC specks. Then, NLRP3 would associate with ASC specks and pro-caspase-1 to form multi-protein complexes of large molecular weights, which was referred as NLRP3 inflammasome. Right part: alternative inflammasome. The priming step is similar to canonical inflammasome (not shown). However, it is signaled by TLR4-caspase-8 that works upstream of NLRP3 and is independent of potassium efflux. At last, in both canonical and alternative inflammasome pathways, NLRP3 inflammasome activation leads to the maturation and secretion of IL-1β and IL-18. Similar to the chloride efflux inhibitors, MCC950 impedes the formation of ASC specks, so MCC950 might correlate with chloride efflux, CLICs, or other targets that act upstream of chloride efflux. Target for MCC950 is denoted as scissors sign. Schematic representation of the domain organization of NLRP3 inflammasome is exhibited in the lower right-hand corner. NLRP3 has a tripartite structure including the LRRs, NACHT, and PYD. ASC is a bipartite protein containing the PYD and CARD. Once being activated, NLRP3 interacts with ASC via homotypic interactions of PYD–PYD while ASC recruits pro-caspase-1 into the complex by the CARD–CARD interactions.
NLRP3 inflammasome is the most widely characterized inflammasome. Accumulating studies showed that aberrant NLRP3 inflammasome activation is pathogenic and can lead to diseases associated with long-term inflammatory process including atherosclerosis [5], type 2 diabetes mellitus [6], and rheumatoid arthritis and gout [7, 8]. Blocking NLRP3 inflammasome activation shows therapeutic effects to the above conditions [9, 10], which strongly indicates its promising roles in treating inflammatory-related diseases.
Several small molecules including isoliquiritigenin, which is a chalcone from Glycyrrhiza uralensis, have been shown to inhibit NLRP3 inflammasome. Moreover, they also inhibit LPS-induced NF-κB (nuclear factor kappa B) activation and decrease the expressions of pro-IL-1β and NLRP3, which shows a multi-target function [11]. Glyburide exhibits an anti-inflammatory effect by inhibiting NLRP3 inflammasome. Nevertheless, other studies show that glyburide does not affect caspase-1 activity, which suggests that it acts upstream of NLRP3 inflammasome activation [12]. Besides, other molecules such as parthenolide, DMSO (dimethyl sulfoxide), and MNS (3,4-methylenedioxy-β-nitrostyrene) exhibit limited and nonspecific anti-inflammatory potency although being capable of inhibiting NLRP3 inflammasome [13].
MCC950 (also known as CP-456,773) is a diarylsulfonylurea-containing compound, which was first identified as an IL-1β inhibitor and therefore classified into CRIDs (cytokine release inhibitory drugs) [14]. Further research of its pharmacological mechanism exhibited that MCC950 impedes IL-1β maturation and release through inhibiting the activation of NLRP3 inflammasome [13, 15]. Today, its therapeutic effects have been widely proved in the animal models with inflammatory-related diseases such as atherosclerosis [16], spontaneous colitis [9], diabetes-mediated cognitive impairment [17], and cholestatic liver injury [18] and even in patients with NLRP3 low penetrance variants [19]. Nonetheless, the molecular target of MCC950 is still to be elucidated. Accordingly, this review focuses on current advances in pharmacological mechanism of MCC950 research to provide insights into its biological/pharmacological mechanism of action.
ACTIVATION OF NLRP3 INFLAMMASOME
Although NLRP3 inflammasome is the most widely characterized one that implicated in inflammatory diseases, its detailed mechanism is still to be elucidated. It has been widely accepted that NLRP3 inflammasome is formed by two steps including priming and NLRP3 inflammasome assembly. In the priming step, inflammatory stimuli such as PAMPs (pathogen-associated molecular patterns) and DAMPs (danger associated molecular patterns) are recognized by TLRs (toll-like receptors), which in turn induce NF-κB-mediated NLRP3, pro-caspase-1, pro-IL-1β, and pro-IL-18 expressions. The second step refers to NLRP3 inflammasome assembly, which is initiated via NLRP3 activation (discussed later). Once being activated, the NLRP3 would associate with ASC (apoptosis-associated speck-like protein containing a CARD) and pro-caspase-1 to form multi-protein complexes of large molecular weights, which was referred as NLRP3 inflammasome. This results in maturation and secretion of some interleukins including IL-1β and IL-18, as well as other mediators of soluble inflammation which are all of potent pro-inflammatory activities [6, 20, 21].
MCC950 IS A HIGHLY SPECIFIC NLRP3 INFLAMMASOME INHIBITOR
A study from Coll et al. [13] exhibited that TLR signaling was not inhibited by MCC950 in LPS-primed monocyte or macrophage as the expressions of NLRP3, pro-IL-1β, and pro-caspase-1 were not impeded. However, the secretions of both caspase-1 and IL-1β were attenuated under MCC950 treatment [22]. It seems that MCC950 does not impact the priming step in NLRP3 inflammasome activation, but rather the step of assembly of NLRP3 inflammasome. What’s more, MCC950 inhibits neither secretion of TNF-α nor activation of AIM2, NLRP1, and NLRC4 inflammasomes [13]. In summary, these results indicate that MCC950 impedes inflammatory cytokine secretion by inhibiting the assembly of NLRP3 inflammasomes specifically.
TARGETS OF MCC950
As discussed above, MCC950 does not abrogate the priming step in NLRP3 inflammasome activation, but rather the step of NLRP3 inflammasome assembly. However, the molecular pathway leading to NLRP3 inflammasome assembly is complex; therefore, the target of MCC950 is still obscure.
Potassium Efflux
A large number of chemically and structurally unrelated PAMPs and DAMPs can activate NLRP3. As a result, NLRP3 is proposed as a common “sensor” for a set of cellular events, rather than a “receptor” that interacts with its activators directly [1, 23]. Subsequent studies revealed that potassium efflux is the converging signal for NLRP3 activation, which is noted as a “common denominator” [24]. It has been demonstrated that increased concentrations of extracellular potassium could attenuate potassium efflux, which also undermine NLRP3 inflammasome activation in macrophages induced by inflammasome-activating agents [24, 25].
Nonetheless, high-potassium treatment could not abolish IL-1β release in human monocyte primed with LPS alone [26], as the one-step NLRP3 inflammasome pathway has been engaged in this situation. This is different from the classical two-step NLRP3 inflammasome signaling, and is potassium efflux independent so termed as “alternative NLRP3 inflammasome pathway” by Hornung et al. [27]. While this alternative pathway can be abrogated by MCC950 [26, 27], it was exhibited that IL-1β secretion was impeded in LPS-primed human monocyte upon MCC950 treatment. Furthermore, Coll et al. [13] demonstrated that MCC950 inhibited IL-1β release without affecting potassium efflux in macrophages. Thus, the action of MCC950 is independent of potassium efflux.
NEK7
NEK7 (NIMA-related serine/threonine kinase 7) is a centrosomal kinase that is required for mitosis [28]. Several studies show that NEK7 is also an NLRP3-binding protein that responds to mediate NLRP3 activation and NLRP3 inflammasome assembly [29, 30]. It has been proposed that NEK7 rather than NLRP3 is the sensor of potassium efflux [31]. Potassium efflux causes NEK7 forming a complex with NLRP3 followed by ASC recruitment and inflammasome assembly [30]. Using co-immunoprecipitation, Xu et al. showed that MCC950 could partially inhibit the interaction between NLRP3 and NEK7 [32].
However, the exact target of MCC950 is still obscure. MCC950 does not work by inhibiting the catalytic activity of NEK7, as it is dispensable for NLRP3 activation [29]. In addition, ROS-dependent phosphorylation of NEK7 could strengthen NEK7–NLRP3 interaction and promote inflammasome activation [30]. Moreover, the ROS scavenger N-acetylcysteine can abolish NEK7 phosphorylation therefore the interaction between NEK7 and NLRP3, and the downstream IL-1β production were impeded. Furthermore, Perera et al. [9] claimed that MCC950 did not impede NEK7 phosphorylation; thus, MCC950 did not work by inhibiting the NEK7–NLRP3 interaction either. However, Xu et al. [32] argued that MCC950 could partially inhibit NEK7–NLRP3 interaction. So, the exact mechanism that how MCC950 interacts with NEK7 needs to be further explored.
ASC
ASC is a bipartite protein containing the N-terminal PYD and the C-terminal CARD [33, 34]. Upon NLRP3 activation, ASC forms an aggregate and appears as a speck through the association between each PYD and CARD. As a result, these specks create multiple potential caspase-1 activation sites that amplify the inflammasome signaling [35, 36].
What’s more, ASC is also an important adaptor protein in NLRP3 inflammasome assembly [34, 37]. In the course of inflammasome assembly, NLRP3 interacts with ASC via homotypic interactions of PYD-PYD while ASC recruits pro-caspase-1 into the complex by the CARD–CARD interactions. Coll et al. [13] exhibit that MCC950 restrains the NLRP3-induced ASC oligomerization rather than their interactions.
Besides, Green et al. [31] proposed that alternative NLRP3 inflammasome forms independent of ASC oligomerization in human peripheral blood mononuclear cells. However, other studies showed that alternative NLRP3 inflammasome pathway in BMCs can still be blocked by MCC950 [26, 27]. These findings imply that the function of MCC950 is not limited to inhibition of ASC oligomerization. Therefore, additional researches are still required to determine the precise molecular mechanism between MCC950 and ASC oligomerizations.
Chloride Efflux
In 2005, Verhoef et al. [38] reported that manipulation of extracellular chloride concentration would affect the ability of ATP-stimulated caspase-1 activation and IL-1β processing. Subsequent reports confirmed that chloride efflux was a signal for ASC oligomerization and played an essential role in NLRP3 inflammasome assembly [31, 39, 40]. What’s more, several other researches showed that CLICs (chloride intracellular channels) were the critical anion channel for Cl− efflux during inflammatory responses [39, 41, 42]. Besides, Tang and colleagues [39] exhibited that although CLICs were not present in NEK7–NLRP3–ASC complex, the association between NEK7 and NLRP3, as well as the subsequent inflammasome assembly, could be suppressed by their inhibitions. They proposed that CLICs might regulate NLRP3 inflammasome assembly via regulating NEK7–NLRP3 interaction. Nonetheless, the exact mechanisms involved in CLICs remain mysterious.
More recently, a novel hypothesis has been raised by Green et al. [31] that facilitating NLRP3 inflammasome assembly needs two events: chloride efflux–dependent ASC speck oligomerization and potassium efflux–dependent activation of caspase-1. As showed by Green and other groups, both ASC speck formation and inflammasome assembly could be interrupted by CLIC inhibition [31, 39, 42]. Furthermore, chloride efflux can occur without potassium efflux. However, the chloride efflux–dependent ASC specks could not activate caspase-1 without potassium efflux. Therefore, these two events are independent during inflammasome assembly but are both indispensable. Moreover, although chloride efflux–dependent ASC speck formation alone did not ensue NLRP3 activation, it does augment inflammasome-dependent responses and is competent for subsequent inflammasome assembly and IL-1β processing [31]. These results may also explain Tang’s findings as discussed above; that is, CLICs might regulate NEK7–NLRP3 interaction indirectly and partially.
In summary, CLIC-dependent chloride efflux plays an important role in ASC speck formation and inflammasome assembly. What’s more, suppression of chloride efflux has no effects on potassium efflux, calcium influx, TNF-α secretion, NLRP3, or caspase-1 mRNA expression, nor AIM2 or NLRC4 inflammasome activation [31, 39, 42].
On the other hand, MCC950 could block ASC oligomerization, but not potassium efflux, TNF-α secretion, AIM2, or NLRC4 inflammasome activation, which has been discussed above. All these evidences indicate that MCC950 might function as an inhibitor of CLICs, or other targets that act upstream of chloride efflux (Fig. 1). To a certain extent, potassium efflux and chloride efflux are two independent events in activating NLRP3 inflammasome. Consequently, the effect of MCC950 on potassium efflux (as another event in the inflammasome pathway) and its subsequent events, such as interaction of NEK7–NLRP3, are limited. What’s more, similar to the chloride efflux inhibitors, MCC950 impedes the formation of ASC specks, which also impede the interaction of NEK7–NLRP3 indirectly. The reason can be explained smoothly by the two contradictory results from Xu and Perera that MCC950 does not inhibit NEK7 phosphorylation but can still partially inhibit NEK7–NLRP3 interaction. This is because MCC950 does not block the phosphorylation of NEK7, but inhibit the NEK7–NLRP3 interaction indirectly.
Question Remain
CLIC family consists of six members (CLICs 1–6) [43]. Impairment in TNF-α secretion was observed in cells with CLIC4 knockout, but not in ones with CLIC1 knockout [42, 44], which indicated a distinct function of each of CLICs in inflammatory response. Hence, to understand the precise interaction between MCC950 and each CLIC, further studies are demanded.
CONCLUSION
Emerging biological roles of chloride efflux in NLRP3 inflammasome assembly give us a new and prompt understanding of the mechanism of inflammasome assembly, which is informative in investigating the target of MCC950 as well. In conclusion, the target of MCC950 might correlate with chloride efflux, CLICs, or other targets that act upstream of chloride efflux.
References
He, Y., H. Hara, and G. Nunez. 2016. Mechanism and regulation of NLRP3 inflammasome activation. Trends in Biochemical Sciences 41: 1012–1021.
Fabio Martinon, K.B. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL. Molecular Cell 10: 417–426.
Guo, H., J.B. Callaway, and J.P. Ting. 2015. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature Medicine 21: 677–687.
Sharma BR, Karki R, Kanneganti TD. 2019. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. European Journal of Immunology.
Zhou, W., C. Chen, Z. Chen, L. Liu, J. Jiang, Z. Wu, M. Zhao, and Y. Chen. 2018. NLRP3: a novel mediator in cardiovascular disease. Journal of Immunology Research 2018: 5702103.
Sepehri, Z., Z. Kiani, M. Afshari, F. Kohan, A. Dalvand, and S. Ghavami. 2017. Inflammasomes and type 2 diabetes: an updated systematic review. Immunology Letters 192: 97–103.
Shen, H.H., Y.X. Yang, X. Meng, X.Y. Luo, X.M. Li, Z.W. Shuai, D.Q. Ye, and H.F. Pan. 2018. NLRP3: a promising therapeutic target for autoimmune diseases. Autoimmunity Reviews 17: 694–702.
Szekanecz, Z., S. Szamosi, G.E. Kovacs, E. Kocsis, and S. Benko. 2019. The NLRP3 inflammasome - interleukin 1 pathway as a therapeutic target in gout. Archives of Biochemistry and Biophysics 670: 82–93.
Perera, A.P., R. Fernando, T. Shinde, R. Gundamaraju, B. Southam, S.S. Sohal, A.A.B. Robertson, K. Schroder, D. Kunde, and R. Eri. 2018. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Scientific Reports 8: 8618.
Coates, B.M., K.L. Staricha, N. Ravindran, C.M. Koch, Y. Cheng, J.M. Davis, D.K. Shumaker, and K.M. Ridge. 2017. Inhibition of the NOD-like receptor protein 3 inflammasome is protective in juvenile influenza A virus infection. Scientific Reports 8: 782.
Honda, H., Y. Nagai, T. Matsunaga, N. Okamoto, Y. Watanabe, K. Tsuneyama, H. Hayashi, I. Fujii, M. Ikutani, Y. Hirai, A. Muraguchi, and K. Takatsu. 2014. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. Journal of Leukocyte Biology 96: 1087–1100.
Lamkanfi, M., J.L. Mueller, A.C. Vitari, S. Misaghi, A. Fedorova, K. Deshayes, W.P. Lee, H.M. Hoffman, and V.M. Dixit. 2009. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. The Journal of Cell Biology. 187: 61–70.
Coll, R.C., A.A. Robertson, J.J. Chae, S.C. Higgins, R. Munoz-Planillo, M.C. Inserra, I. Vetter, L.S. Dungan, B.G. Monks, A. Stutz, D.E. Croker, M.S. Butler, M. Haneklaus, C.E. Sutton, G. Nunez, E. Latz, D.L. Kastner, K.H. Mills, S.L. Masters, K. Schroder, M.A. Cooper, and L.A. O'Neill. 2015. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine 21: 248–255.
Perregaux, D.G., P. McNiff, R. Laliberte, N. Hawryluk, H. Peurano, E. Stam, J. Eggler, R. Griffiths, M.A. Dombroski, and C.A. Gabel. 2001. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. The Journal of Pharmacology and Experimental Therapeutics. 299: 187–197.
Primiano, M.J., and B.A. Lefker. 2016. Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary. Inflammation. 197: 2421–2433.
van der Heijden, T., E. Kritikou, W. Venema, J. van Duijn, P.J. van Santbrink, B. Slutter, A.C. Foks, I. Bot, and J. Kuiper. 2017. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arteriosclerosis, Thrombosis, and Vascular Biology 37: 1457–1461.
Ward, R., W. Li, Y. Abdul, L. Jackson, G. Dong, S. Jamil, J. Filosa, S.C. Fagan, and A. Ergul. 2019. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacological Research 142: 237–250.
Qu, J., Z. Yuan, G. Wang, X. Wang, and K. Li. 2019. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. International Immunopharmacology 70: 147–155.
Schuh, E., C.J. Gross, D. Wagner, M. Schluter, O. Gross, and T. Kumpfel. 2019. MCC950 blocks enhanced interleukin-1beta production in patients with NLRP3 low penetrance variants. Clinical Immunology 203: 45–52.
Schroder, K., and J. Tschopp. 2010. The inflammasomes. Cell. 140: 821–832.
Shao, B.Z., Z.Q. Xu, B.Z. Han, D.F. Su, and C. Liu. 2015. NLRP3 inflammasome and its inhibitors: a review. Frontiers in Pharmacology 6: 262.
Zhang, Y., X. Lv, Z. Hu, X. Ye, X. Zheng, Y. Ding, P. Xie, and Q. Liu. 2017. Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death & Disease 8: e2941.
Gaidt, M.M., and V. Hornung. 2018. The NLRP3 inflammasome renders cell death pro-inflammatory. Journal of Molecular Biology 430: 133–141.
Munoz-Planillo, R., P. Kuffa, G. Martinez-Colon, B.L. Smith, T.M. Rajendiran, and G. Nunez. 2013. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 38: 1142–1153.
Petrilli, V., S. Papin, C. Dostert, A. Mayor, F. Martinon, and J. Tschopp. 2007. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation 14: 1583–1589.
Gov, L., C.A. Schneider, T.S. Lima, W. Pandori, and M.B. Lodoen. 2017. NLRP3 and potassium efflux drive rapid IL-1beta release from primary human monocytes during Toxoplasma gondii infection. Journal of Immunology (Baltimore, Md. : 1950) 199: 2855–2864.
Gaidt, M.M., T.S. Ebert, D. Chauhan, T. Schmidt, J.L. Schmid-Burgk, F. Rapino, A.A. Robertson, M.A. Cooper, T. Graf, and V. Hornung. 2016. Human monocytes engage an alternative inflammasome pathway. Immunity. 44: 833–846.
Fry, A.M., L. O'Regan, S.R. Sabir, and R. Bayliss. 2012. Cell cycle regulation by the NEK family of protein kinases. Journal of Cell Science 125: 4423–4433.
He, Y., M.Y. Zeng, D. Yang, B. Motro, and G. Nunez. 2016. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 530: 354–357.
Shi, H., Y. Wang, X. Li, X. Zhan, M. Tang, M. Fina, L. Su, D. Pratt, C.H. Bu, S. Hildebrand, S. Lyon, L. Scott, J. Quan, Q. Sun, J. Russell, S. Arnett, P. Jurek, D. Chen, V.V. Kravchenko, J.C. Mathison, E.M. Moresco, N.L. Monson, R.J. Ulevitch, and B. Beutler. 2016. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nature Immunology 17: 250–258.
Green, J.P., S. Yu, F. Martin-Sanchez, P. Pelegrin, G. Lopez-Castejon, C.B. Lawrence, and D. Brough. 2018. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proceedings of the National Academy of Sciences of the United States of America 115: E9371–E9E80.
Xu, K.Y., C.Y. Wu, S. Tong, P. Xiong, and S.H. Wang. 2018. The selective Nlrp3 inflammasome inhibitor Mcc950 attenuates lung ischemia-reperfusion injury. Biochemical and Biophysical Research Communications 503: 3031–3037.
Masumoto, J., S. Taniguchi, K. Ayukawa, H. Sarvotham, T. Kishino, N. Niikawa, E. Hidaka, T. Katsuyama, T. Higuchi, and J. Sagara. 1999. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. The Journal of Biological Chemistry. 274: 33835–33838.
Gumucio, D.L., A. Diaz, P. Schaner, N. Richards, C. Babcock, M. Schaller, and T. Cesena. 2002. Fire and ICE: the role of pyrin domain-containing proteins in inflammation and apoptosis. Clinical and Experimental Rheumatology 20: S45–S53.
Awad, F., E. Assrawi, C. Louvrier, C. Jumeau, S. Georgin-Lavialle, G. Grateau, S. Amselem, I. Giurgea, and S.A. Karabina. 2018. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacology & Therapeutics 187: 133–149.
Dick, M.S., L. Sborgi, S. Ruhl, S. Hiller, and P. Broz. 2016. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nature Communications 7: 11929.
Richards, N., P. Schaner, A. Diaz, J. Stuckey, E. Shelden, A. Wadhwa, and D.L. Gumucio. 2001. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. The Journal of Biological Chemistry. 276: 39320–39329.
Verhoef, P.A., S.B. Kertesy, K. Lundberg, J.M. Kahlenberg, and G.R. Dubyak. 2005. Inhibitory effects of chloride on the activation of caspase-1, IL-1 secretion, and cytolysis by the P2X7 receptor. The Journal of Immunology. 175: 7623–7634.
Tang, T., X. Lang, C. Xu, X. Wang, T. Gong, Y. Yang, J. Cui, L. Bai, J. Wang, W. Jiang, and R. Zhou. 2017. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nature Communications 8: 202.
Daniels, M.J., J. Rivers-Auty, T. Schilling, N.G. Spencer, W. Watremez, V. Fasolino, S.J. Booth, C.S. White, A.G. Baldwin, S. Freeman, R. Wong, C. Latta, S. Yu, J. Jackson, N. Fischer, V. Koziel, T. Pillot, J. Bagnall, S.M. Allan, P. Paszek, J. Galea, M.K. Harte, C. Eder, C.B. Lawrence, and D. Brough. 2016. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nature Communications 7: 12504.
Laliberte, R.E., D.G. Perregaux, L.R. Hoth, P.J. Rosner, C.K. Jordan, K.M. Peese, J.F. Eggler, M.A. Dombroski, K.F. Geoghegan, and C.A. Gabel. 2003. Glutathione s-transferase omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1beta posttranslational processing. The Journal of Biological Chemistry. 278: 16567–16578.
Domingo-Fernandez, R., R.C. Coll, J. Kearney, S. Breit, and L.A.J. O'Neill. 2017. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. The Journal of Biological Chemistry. 292: 12077–12087.
Argenzio, E., and W.H. Moolenaar. 2016. Emerging biological roles of Cl- intracellular channel proteins. Journal of Cell Science 129: 4165–4174.
He, G., Y. Ma, S.Y. Chou, H. Li, C. Yang, J.Z. Chuang, C.H. Sung, and A. Ding. 2011. Role of CLIC4 in the host innate responses to bacterial lipopolysaccharide. European Journal of Immunology 41: 1221–1230.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81873130, No. 81573733, No. 81804025), Tianjin Municipal Natural Science Foundation (No. 18JCYBJC94500), Scientific and Technological Research Program of Tianjin Municipal Education Commission (No. 2017KJ164), and 2017 Annual Graduate Students Innovation Fund (School of Integrative Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China; No. CXJJLX201701).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, D., Chen, Y., Sun, Y. et al. Target of MCC950 in Inhibition of NLRP3 Inflammasome Activation: a Literature Review. Inflammation 43, 17–23 (2020). https://doi.org/10.1007/s10753-019-01098-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01098-8