
Abstract
Sepsis progression is linked to the imbalance between reactive oxygen species and antioxidant enzymes. Sepsis affects multiple organs, but when associated with a chronic inflammatory disease, such as obesity, it may be exacerbated. We hypothesized that obesity could aggravate the oxidative damage to peripheral organs of rats submitted to an animal model of sepsis. Male Wistar rats aged 8 weeks received hypercaloric nutrition for 2 months to induce obesity. Sepsis was induced by cecal ligation and puncture (CLP) procedure, and sham-operated rats were considered as control group. The experimental groups were divided into sham + eutrophic, sham + obese, CLP + eutrophic, and CLP + obese. Twelve and 24 h after surgery, oxidative damage to lipids and proteins and superoxide dismutase (SOD) and catalase (CAT) activities were evaluated in the liver, lung, kidney, and heart. The data indicate that obese rats subjected to sepsis present oxidative stress mainly in the lung and liver. This alteration reflected an oxidative damage to lipids and proteins and an imbalance of SOD and CAT levels, especially 24 h after sepsis. It follows that obesity due to its pro-inflammatory phenotype can aggravate sepsis-induced damage in peripheral organs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Sepsis is a major cause of mortality in noncardiac intensive care units and is an important cause of death in both developed and underdeveloped countries [1–3]. This syndrome is a severe clinical condition as consequence of a systemic inflammatory state with release of endogenous mediators and the triggering of inflammatory cascades due to the occurrence of a suspected or diagnosed infection [4, 5].
Activation of inflammatory cascades through cytokine stimulation of immune and endothelial cells results in increased production of reactive oxygen species (ROS) [3, 6]. Evidences suggest that an excessive ROS production added to an impairment in its degradation by cellular antioxidants, a process called oxidative stress, can damage lipids, proteins, carbohydrates, and nucleic acids, which contributes to sepsis severity and its progression, leading to organ dysfunction and failure, especially in the liver, lungs, heart, and kidneys [7–9].
Oxidative stress is related not only to sepsis but also to various conditions, such as obesity [1, 10]. Obesity is a chronic metabolic disease characterized by excessive fat accumulation and adipose tissue overgrowth [2, 11]. This condition compromises the body’s ability to adapt to critical illness and is associated with increased morbidity and all-cause mortality. It is known that obese individuals exhibit elevated levels of ROS and antioxidant defense impairment [12, 13], altered inflammatory response and immune cell function [14], have higher risk to develop sepsis and septic shock, as well as higher mortality rates than eutrophic individuals [15].
Previously, we evaluated the susceptibility to brain damage after sepsis in obese rats treated with a hypercaloric diet, indicating that obesity, due to its pro-inflammatory phenotype, can aggravate or accelerate sepsis-induced damage in rat brain [16]. We hypothesized that obesity could also aggravate the oxidative damage to peripheral organs of rats submitted to an animal model of sepsis. To test our hypothesis, we evaluated lipid and protein oxidative damage and antioxidant enzyme activities in the liver, lung, heart, and kidney in animals subjected to a clinically relevant animal model of sepsis and to prolonged diet-induced obesity.
MATERIALS AND METHOD
Animals
Male Wistar rats (250–300 g), aged 8 weeks, were housed five to a standard cage in a controlled animal facility, with a 12-h light/dark cycle (lights on at 7:00 a.m.). The animals had free access to tap water and food. All studies were performed in accordance with the National Institutes of Health Guidelines and with the approval of the Animal Care and Experimentation Committee of UNISUL (protocol number 13.026.4.03.IV), Brazil.
Obesity Induction
For 2 months, the animals received hypercaloric diet previously standardized [17], containing by weight 19 % protein, 47 % carbohydrate, 16 % fat, 3 % cellulose, 5 % of vitamins and minerals to 4.79 kcal/g (obese group) or feed normocaloric Nuvilab CR-1 (eutrophic group). The ingredients were blended and milled in the form of pellets, as follows: 15 g of feed normocaloric Nuvilab CR-1 (3.78 kcal/g), 10 g of roasted peanuts (5.95 kcal/g), 10 g of milk chocolate (5.4 kcal/g), and 5 g of cornstarch wafer (4.25 kcal/g). During the experiment, the animals were weighed once a week. Lee index was used to calculate the evolution of the body mass, which is based on calculating the cubic root of body mass (g) divided by naso-anal length (cm) [18]. The food was provided daily, and the dietary intake was estimated by the difference between the offered food and leftovers.
Sepsis Induction—CLP
After the obesity induction period, sepsis was produced by cecal ligation and puncture (CLP), as previously described [19]. Briefly, animals were anesthetized intraperitoneally with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). Under aseptic conditions, a 3-cm midline laparotomy was performed to expose the cecum and adjoining intestine. The cecum was tightly ligated with a 3.0 silk suture at its base (below the ileocecal valve), perforated once with a 14-gauge needle, squeezed gently to extrude a small amount of feces through the perforation site, and was then returned to the peritoneal cavity, and the laparotomy was closed with 4.0 silk sutures. Animals were resuscitated with regular saline (50 ml/kg) subcutaneously (s.c.) immediately after and 12 h after CLP. All animals were returned to their cages with free access to food and water. In the sham-operated group, the rats were submitted to all surgical procedures but the cecum was neither ligated nor perforated. To minimize variability between different experiments, the same investigator always performed the CLP procedure. The groups were observed after CLP for the presence of signs of infection (piloerection, lethargy, tachypnea, and weight loss).
Experimental Groups and Sample Obtention
Animals were randomly divided into four groups (n = 15): (1) sham + eutrophic, (2) sham + obese, (3) CLP + eutrophic, and (4) CLP + obese. In 12 and 24 h after the surgery procedure (CLP or sham), all rats were euthanized by decapitation. The liver, lung, heart, and kidney were isolated by hand dissection using appropriate surgical instruments and stored at −80 °C for biochemical analyses.
Biochemical Parameters
Thiobarbituric Acid Reactive Species Formation
Lipid peroxidation was measured by formation of thiobarbituric acid (TBA) reactive substances (TBARS) [20]. After organ dissection, samples were washed with PBS, harvested, and lysed. TBARS, obtained by acid hydrolysis of 1,1,3,3-tetraethoxypropane (TEP), were used as the standard for the quantification of TBARS. TBA 0.67 % was added to each tube and vortexed. The reaction mixture was incubated at 90 °C for 20 min, and the reaction was stopped by placing samples on ice. The optical density of each solution was measured in a spectrophotometer at 535 nm. Data were expressed as nmol of malondialdehyde (MDA) equivalents per milligram of protein.
Carbonyl Protein Formation
Carbonyl protein content was measured in brain homogenates using 2,4-dinitrophenylhydrazine (DNPH) in a spectrophotometric assay [21]. Briefly, tissue samples were sonicated in an ice-cold old homogenization buffer containing phosphatase and protease inhibitors (200 nm calyculin, 10 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM sodium orthovanadate, and 1 μM microcystin-LR) and centrifuged at 1000×g for 15 min to sediment insoluble material. Three hundred microliter aliquots of the supernatant containing 0.7–1.5 mg of protein were treated with 300 μl of 10 mM DNPH, dissolved in 2 M HCl, and compared with 2 M HCl alone (reagent blank). Samples were incubated for 1 h at room temperature in the dark, stirred every 10 min, precipitated with trichloroacetic acid (final concentration of 20 %), and centrifuged at 16,000×g at 4 °C for 15 min. The pellet was washed three times with 1 ml of ethanol/ethyl acetate (1:1 v/v). Each time, the pellet was lightly vortexed and left exposed to the washing solution for 10 min before centrifugation (16,000×g for 5 min). The final pellet was dissolved in 1 ml of 6 M guanidine in 10 mM phosphate buffer-trifluoroacetic acid, pH 2.3, and the insoluble material was removed by centrifugation at 16,000×g for 5 min. Absorbance was recorded in a spectrophotometer at 370 nm for both DNPH-treated and HCl-treated samples. Carbonyl protein levels were expressed as nmoles of carbonyl per milligram of protein.
Superoxide Dismutase Activity
Superoxide dismutase (SOD) estimation was performed based on its ability to spontaneously inhibit oxidation of adrenaline to adrenochrome [22]. An amount of 2.78 ml of sodium carbonate buffer (0.05 mM; pH 10.2), 100 μl of EDTA (1.0 mM), and 20 μl of the supernatant or sucrose (blank) were incubated at 30 °C for 45 min. Thereafter, we added 100 μl of adrenaline solution (9.0 mM) to initiate the reaction. Temperature was maintained at 30 °C throughout the assay procedure. The change in the absorbance was recorded at 480 nm for 8 min. One unit of SOD produced approximately 50 % of auto-oxidation of adrenaline. Results were expressed as units/milligram of protein.
Catalase Activity
We measured catalase (CAT) activity by the method that employs hydrogen peroxide (H2O2) to generate H2O and O2 [23]. Tissue samples were sonicated in 50 mmol/l phosphate buffer (pH 7.0), and the resulting suspension was centrifuged at 3000×g for 10 min. A sample aliquot (20 μl) was added to 980 μl of the substrate mixture, and the substrate mixture contained 0.3 ml of hydrogen peroxide in 50 ml of 0.05 M phosphate buffer (pH 7.0). The initial and final absorbance were recorded at 240 nm after 1 and 6 min, respectively. A standard curve was established using purified catalase (Sigma, MO, USA) under identical conditions.
Protein Determination
All biochemical measures were normalized to the protein content with bovine albumin as standard [24].
Statistical Analysis
Data were analyzed using the Statistical Package for the Social Sciences software (SPSS, Chicago, IL, USA). Data are expressed as mean ± SD, and differences among experimental groups were determined by Student’s t test or one-way analysis of variance (ANOVA), followed by Tukey post hoc. In all comparisons, statistical significance was set at p < 0.05.
RESULTS
Table 1 illustrates the nutritional status and food intake of the animals. Animals subjected to hypercaloric diet gain weight compared to animals treated with normocaloric diet, which promoted a significant increase in the Lee index. Differences in the naso-anal length and food intake among groups were not observed, as previously demonstrated [16].
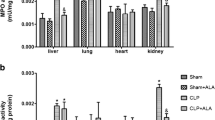
Figures 1 and 2 illustrate the MDA equivalent levels, as a lipid peroxidation marker, and carbonyl protein content, as a protein oxidative damage parameter, in the liver, lung, kidney, and heart of the rats, 12 and 24 h after CLP procedure. Obesity, per se, significantly stimulated lipid peroxidation in the liver at 12 h after CLP surgery (Fig. 1a), and this condition lasted for 24 h (Fig. 1b). In addition, septic obese animals presented higher levels of lipid peroxidation in the liver and lungs, but it remained elevated until 24 h after surgery only in the lung. Obesity did not influence lipid peroxidation in the kidney and heart.

Oxidative damage to lipids in the liver, lung, kidney, and heart after sepsis induction in rats submitted to obesity. For 2 months, Wistar rats with 60 days received hypercaloric nutrition to induce obesity. After this period, sepsis was induced by CLP and TBARS levels as a lipid peroxidation marker was measured in 12 (a) and 24 h (b) after CLP (n = 5–7 animals per group). Results are reported as mean ± SD, ANOVA + Tukey post hoc. *p < 0.05 difference from the sham + eutrophic; % p < 0.05 difference from the sham + obese; # p < 0.05 difference from the CLP + eutrophic. TBARS thiobarbituric acid reactive species, CLP cecal ligation and puncture.

Oxidative damage to proteins in the liver, lung, kidney, and heart after sepsis induction in rats submitted to obesity. For 2 months, Wistar rats with 60 days received hypercaloric nutrition to induce obesity. After this period, sepsis was induced by CLP and carbonyl protein levels was measured in 12 (a) and 24 h (b) after CLP (n = 5–7 animals per group). Results are reported as mean ± SD, ANOVA + Tukey post hoc. *p < 0.05 difference from the sham + eutrophic; % p < 0.05 difference from the sham + obese; # p < 0.05 difference from the CLP + eutrophic. CLP cecal ligation and puncture.
Regarding protein carbonylation levels, obesity led to a significant increase in protein oxidation in the lung 12 h after sepsis induction (Fig. 2a). When sepsis was associated with obesity, excessive adiposity potentiated the protein damage in the lung until 24 h after CLP procedure (Fig. 2b). Obesity did not have an effect on susceptibility to protein damage occurrence in the liver, kidney, and heart (Fig. 2a, b).
Figure 3 elucidates the SOD activity as antioxidant defense capacity parameters in the liver, lung, heart, and kidney of the rats, 12 and 24 h after CLP procedure. According to Fig. 3a, obesity significantly intensified the reduction in SOD activity levels in the liver and kidney of septic animals. In the first 12 h, obesity did not affect SOD activity in the lung; however, septic obese animals showed a reduction in SOD activity in pulmonary tissue, 24 h after surgery (Fig. 3b).

SOD activity in the liver, lung, kidney, and heart after sepsis induction in rats submitted to obesity. For 2 months, Wistar rats with 60 days received hypercaloric nutrition to induce obesity. After this period, sepsis was induced by CLP and SOD levels was measured in 12 (a) and 24 h (b) after CLP (n = 5–7 animals per group). Results are reported as mean ± SD, ANOVA + Tukey post hoc. *p < 0.05 difference from the sham + eutrophic; % p < 0.05 difference from the sham + obese; # p < 0.05 difference from the CLP + eutrophic. SOD superoxide dismutase, CLP cecal ligation and puncture.
CAT activity levels at 12 h after induction are demonstrated in Fig. 4a. Obesity promoted an increased reduction in CAT activity level in the lungs of septic animals, although this disturbance did not remain until 24 h after CLP procedure. Indeed, after 24 h, CAT activity was not significantly altered in any of the four organs evaluated (Fig. 4b).

CAT activity in the liver, lung, kidney, and heart after sepsis induction in rats submitted to obesity. For 2 months, Wistar rats with 60 days received hypercaloric nutrition to induce obesity. After this period, sepsis was induced by CLP and CAT levels was measured in 12 (a) and 24 h (b) after CLP (n = 5–7 animals per group). Results are reported as mean ± SD, ANOVA + Tukey post hoc. *p < 0.05 difference from the sham + eutrophic; % p < 0.05 difference from the sham + obese; # p < 0.05 difference from the CLP + eutrophic. CAT catalase, CLP cecal ligation and puncture.
DISCUSSION
In the present study, we demonstrated the influence of obesity on the organs damaged after sepsis in animals. The data indicate that rats treated with a hypercaloric diet and subjected to sepsis have an enhanced oxidative damage in lipids and proteins and imbalanced antioxidant defenses, especially in the lung and liver. Our findings suggest that obese animals are more vulnerable to the deleterious effects of sepsis on peripheral organs.
Obesity, per se, generates inflammation owing to its phenotype and metabolic consequences, such as insulin resistance. In this scenario, an additional inflammatory stimulus results in a situation called “second hit,” which leads to an exaggerated inflammatory response and implicates obesity as a risk factor for sepsis-induced multiple organ dysfunction [16, 25]. We induced obesity through a hypercaloric diet consumption, as previously established by Estadella et al. [17]. This hypercaloric diet promotes obesity and enhances lipid content in the liver, although the hepatic wet weigh is not affected.
Liver is a crucial metabolic organ and plays an important role for fatty acids and cholesterol metabolism [26], including fatty acid and triglyceride synthesis and storage, apolipoprotein assembly and secretion, lipolysis, and fatty acid oxidation. The endoplasmic reticulum (ER) and mitochondria are the major sites wherein these aspects take place [27, 28], but under energy and nutrient excess conditions, such as obesity, the organism cannot maintain homeostasis [26, 29]. Hypercaloric diet consumption, and consequently increased intrahepatic levels of lipids, can disrupt ER and mitochondria, leading to a stressful state of these organelles and loss of metabolic functions [30]. This diet pattern induces mitochondrial dysfunction and ER stress in the liver and other tissues, in addition to hepatic steatosis, insulin resistance, and type 2 diabetes [31].
In our study, we found that obesity did not influence some of the biochemical parameters assessed 12 h after sepsis induction, including lipid peroxidation in the kidney and heart, protein carbonylation in the liver and kidney, as well as SOD antioxidant activity in the lung. Thus, obese animals subjected to sepsis induction showed no further damage to these organs, considering the biochemical markers adopted in this research.
Evidence suggests that hypercaloric diet induces changes in renal lipid metabolism due to a local imbalance between lipogenesis and lipolysis, as well as systemic metabolic abnormality, lipid accumulation, and renal injury, which causes obesity-associated renal disease [32, 33]. This scenario is preceded by endothelial dysfunction and hypertension, both generated by oxidative stress [34, 35]. Besides, this model promotes changes in lipid and glucose metabolism and it is usual to observe an increase in plasma glucose and insulin levels, lipoprotein cholesterol carriers, and triglycerides. Together, these changes may indicate the presence of insulin resistance and oxidative stress [36, 37].
The literature clearly establishes the relationship between cellular damage and acute toxicity due to elevated levels of glucose, which affects mainly cells with glucose uptake pathways not dependent on insulin, such as hepatocytes, kidney cells, and endothelial cells [38, 39]. However, insulin-dependent cells, such as cardiac cells, can also be affected when a loss of function of this hormone exists, considering that there will be less receptor translocation for membrane cell and glucose uptake, which induces the use of free fatty acids as an energy source by the myocardium. Hence, this pattern results in ROS generation and propensity to fatty acid peroxidation [38]. Furthermore, the literature suggests that hyperglycemia causes severe oxidative damage to mitochondria, as well as other metabolic and immunological effects, including alterations in the circulating lipid profile, endothelial dysfunction, and decreased neutrophil function, which may negatively affect organ function.
Due to the toxical cellular effect, mitochondrial damage, and oxidative stress, glycemic control in critical illness, including sepsis, has been considered an important prognostic criterion. The dietary model of obesity induction through hypercaloric diet causes changes in glucose metabolism, as demonstrated above and supported by literature. In other words, both sepsis and the diet can stimulate oxidative damage by hyperglycemia. In our study, we did not evaluate diagnostic parameters for these particular alterations but we speculate that the results not influenced by obesity may be related to the factors mentioned above.
Since ER and mitochondria are responsible for lipid metabolism and oxidation, these are important sources of endogenous intracellular ROS, but under an impaired function condition, an increased ROS emission and oxidative stress occur [30]. Reactive oxidative species are crucial, when produced at physiological levels, as they mediate vital cellular processes and signaling networks; however, appropriate antioxidant levels are necessary to prevent cellular damage and maintain ROS’s beneficial effects and organic balance [13]. ROS can stimulate lipid peroxidation within the cell through polyunsaturated fatty acid (PUFA) damage and MDA formation. MDA have longer half-life than ROS, and it is able to spread and reach distant intracellular and extracellular targets, thereby amplifying the effects of oxidative stress [40].
In our study, we found that 24 h after sepsis induction, oxidative damage to lipids and proteins remained significantly higher in the lung of animals with sepsis and obesity. These data indicate that diet-induced obesity aggravates the oxidative damage promoted by sepsis mainly in the lung. Male Kunming mice subjected to CLP procedure showed increased production of pro-inflammatory markers and significant infiltration of inflammatory cells, extensive thickening of the alveolar wall, and demolished structure of pulmonary alveoli, 24 h after induction [41]. Sepsis induction by CLP in male adult Sprague-Dawley rats caused thickened and congested alveolar walls, edema fluid, and leukocyte infiltration, which is consistent with acute lung injury [42]. Mice treated with a high-fat diet for 3 weeks and subjected to sepsis induction by CLP showed enhanced inflammatory markers, such as neutrophil infiltrate, in the liver and lung, and these markers were higher in the pulmonary tissue [43]. Evidence suggests that one mechanism of lung injury is the enhanced ROS production from polymorphonuclear cells, as well as a severely impaired antioxidant system in this organ [44].
Regarding antioxidant activity, we demonstrated that obesity exacerbated sepsis-induced effects in reducing antioxidant enzymes levels. Septic obese animals presented, 12 h after induction, a decrease in SOD activity in the liver and kidney, while CAT activity was reduced in the lung. Concerning the antioxidant capacity after 24 h, we observed decreased levels of SOD activity in the lungs of animals subjected to obesity and sepsis but CAT activity levels were normalized in all organs. These results indicate the role of obesity in upregulating sepsis-induced effects in the antioxidant capacity, mostly in the liver and lung.
Decreased antioxidant activity is an expected condition in this rodent model of sepsis induction [45, 46], and multiple organ dysfunctions in sepsis are associated with increased ROS production and antioxidant depletion, leading to oxidative stress [47]. Oxidative stress in obese (ob/ob) or lean mice was observed by Xiao et al. [48] in which it was determined the existence of an oxidative stress in ob/ob animal lungs at baseline and it was exacerbated by cecal ligation and puncture. Since oxidative damage was enhanced in the lung, and SOD expression is enhanced in this organ [49] while CAT expression is elevated in the liver and kidney [50], it is possible that these antioxidant enzyme activities were reduced in this organ due to its elevated requirement and site of expression.
In summary, this study indicates that obese animals are more susceptible to the deleterious effects of sepsis on the peripheral organs, due to the oxidative damage and the low enzymatic antioxidant levels and will become a major focus of our future investigations.
References
Karapetsa, M., M. Pitsika, N. Goutzourelas, D. Stagos, A.T. Becker, et al. 2013. Oxidative status in ICU patients with septic shock. Food Chemistry and Toxicology 61: 106–111.
Kolyva, A.S., V. Zolota, D. Mpatsoulis, G. Skroubis, E.E. Solomou, I.G. Habeos, et al. 2014. The role of obesity in the immune response during sepsis. Nutrition and Diabetes 4(137): 1–7.
Rocha, M., R. Herance, S. Rovira, A. Hernández-Mijares, and V.M. Victor. 2012. Mitochondrial dysfunction and antioxidant therapy in sepsis. Infectious Disorder Drug Targets 12(2): 161–178.
Aziz, M., A. Jacob, W.L. Yang, A. Matsuda, and P. Wang. 2013. Current trends in inflammatory and immunomodulatory mediators in sepsis. Journal Leukocytes Biology 93(3): 329–342.
Xiao, Z., C. Wilson, H.L. Robertson, D.J. Roberts, C.G. Ball, C.N. Jenne, et al. 2015. Inflammatory mediators in intra-abdominal sepsis or injury—a scoping review. Critical Care 19: 373.
Barichello, T., J.J. Fortunato, A.M. Vitali, G. Feier, A. Reinke, J.C. Moreira, et al. 2006. Oxidative variables in the rat brain after sepsis induced by cecal ligation and perforation. Critical Care Medicine 34(3): 886–889.
Andrades, M., C. Ritter, M.R. de Oliveira, E.L. Streck, J.C. Fonseca Moreira, and F. Dal-Pizzol. 2011. Antioxidant treatment reverses organ failure in rat model of sepsis: role of antioxidant enzymes imbalance, neutrophil infiltration, and oxidative stress. Journal of Surgical Research 167(2): e307–e313.
Ritter, C., M. Andrades, M.L. Frota Júnior, F. Bonatto, R.A. Pinho, M. Polydoro, et al. 2003. Oxidative parameters and mortality in sepsis induced by cecal ligation and perforation. Intensive Care Medicine 29(10): 1782–1789.
Zapelini, P.H., G.T. Rezin, M.R. Cardoso, C. Ritter, F. Klamt, J.C. Moreira, et al. 2008. Antioxidant treatment reverses mitochondrial dysfunction in a sepsis animal model. Mitochondrion 8(3): 211–218.
Matsuda, M., and I. Shimomura. 2013. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obesity Research and Clinical Practice 7(5): e330–e341.
Murdolo, G.M., F. Piroddi, C. Luchetti, B. Tortoioli, C. Canonico, C. Zerbinati, et al. 2013. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 95(3): 585–594.
Genoni, G., F. Prodam, A. Marolda, E. Giglione, I. Demarchi, S. Bellone, et al. 2014. Obesity and infection: two sides of one coin. European Journal of Pediatrics 173(1): 25–32.
Huang, C.J., M.J. McAllister, A.L. Slusher, H.E. Webb, J.T. Mock, and E.O. Acevedo. 2015. Obesity-Related Oxidative Stress: the Impact of Physical Activity and Diet Manipulation. Sports Medicine – Open 1(1): 32.
Milner, J.J., and M.A. Beck. 2012. The impact of obesity on the immune response to infection. The Proceedings of the Nutritional Society 71(2): 298–306.
Wurzinger, B., M.W. Dünser, C. Wohlmut, M.C. Deutinger, H. Ulmer, C. Torgersen, et al. 2010. The association between body-mass index and patient outcome in septic shock: a retrospective cohort study. Wiener Klinische Wochenschrift 122(1-2): 31–36.
Vieira, A.A., M. Michels, D. Florentino, D.Z. Nascimento, G.T. Rezin, D.D. Leffa, et al. 2015. Obesity promotes oxidative stress and exacerbates sepsis-induced brain damage. Current Neurovascular Research 12(2): 147–154.
Estadella, D., L.M. Oyama, A.R. Damaso, E.B. Ribeiro, and C.M. Oller Do Nascimento. 2004. Effect of palatable hyperlipidic diet on lipid metabolism of sedentary and exercised rats. Nutrition 20(2): 218–224.
Bernardis, L.L., and B.D. Patterson. 1968. Correlation between ‘Lee index’ and carcass fat content in weanling and adult female rats with hypothalamic lesions. Journal of Endocrinology 40(4): 527–528.
Hubbard, W.J., M. Choudhry, M.G. Schwacha, J.D. Kerby, L.W. Rue, K.I. Bland, et al. 2005. Cecal ligation and puncture. Shock 24(Suppl 1): 52–57.
Draper, H.H., and M. Hadley. 1990. Malondialdehyde determination as índex of lipid peroxidation. Methods Enzymology 186: 421–431.
Levine, R.L., D. Garland, C.N. Oliver, A. Amici, I. Climent, A.G. Lenz, et al. 1990. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymology 186: 464–478.
Bannister, J.V., and L. Caberese. 1987. Assays for superoxide dismutase. Methods Biological Analytical 32: 231–279.
Aebi, H. 1984. Catalase in vitro. Methods Enzymology 105: 121–126.
Lowry, O.H., A.L. Rosebrough, A.L. Farr, and R.J. Randal. 1951. Protein measurement with the Folin phenol reagent. Journal of Biological and Chemistry 193(1): 265–275.
Vachharajani, V. 2008. Influence of obesity on sepsis. Pathophysiology 15(2): 123–134.
Zhou, L., and F. Liu. 2010. Autophagy: roles in obesity-induced ER stress and adiponectin downregulation in adipocytes. Autophagy 6(8): 1196–1197.
Wang, S., and R.J. Kaufman. 2014. How does protein misfolding in the endoplasmic reticulum affect lipid metabolism in the liver? Current Opinion Lipidology 25(2): 125–132.
Fu, S., S.M. Watkins, and G.S. Hotamisligil. 2012. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metabolism 15(5): 623–634.
Yang, L., P. Li, S. Fu, E.S. Calay, and G.S. Hotamisligil. 2010. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metabolism 11(6): 467–478.
Chang, Y.C., S.W. Hee, M.L. Hsieh, Y.M. Jeng, and L.M. Chuang. 2015. The Role of Organelle Stresses in Diabetes Mellitus and Obesity: Implication for Treatment. Analytical Cellular Pathology (Amst) 2015: 972891.
Guo, B., and Z. Li. 2014. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Frontiers in Genetics 5: 242.
Kume, S., T. Uzu, A. Shin-ichi, S. Toshiro, I. Keiji, C.K. Masami, et al. 2008. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. Journal of American Society of Nephrology 18(10): 2715–2723.
Roza, N.A., L.F. Possignolo, A.C. Palanch, and J.A. Gontijo. 2016. Effect of long-term high-fat diet intake on peripheral insulin sensibility, blood pressure, and renal function in female rats. Food Nutrition Research 60: 28536.
Knight, S.F., J.E. Quigley, J. Yuan, S.S. Roy, A. Elmarakby, and J.D. Imig. 2008. Endothelial dysfunction and the development of renal injury in spontaneously hypertensive rats fed a high-fat diet. Hypertension 51(2): 352–359.
Pinhal, C.S., A. Lopes, D.B. Torres, S.L. Felisbino, J.A. Rocha Gontijo, and P.A. Boer. 2013. Time-course morphological and functional disorders of the kidney induced by long-term high-fat diet intake in female rats. Nephrology Dialysis and Transplantation 28(10): 2464–2476.
Maithilikarpagaselvi, N., M.G. Sridhar, R.P. Swaminathan, and R. Sripradha. 2016. Preventive effect of curcumin on inflammation, oxidative stress and insulin resistance in high-fat fed obese rats. Journal of Complementary and Integrative Medicine 13(2): 137–143.
Rani, V., G. Deep, R.K. Singh, K. Palle, and U.C. Yadav. 2016. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Science 148: 83–93.
Abraham, E., and M. Singer. 2007. Mechanisms of sepsis-induced organ dysfunction. Crit Care Medicine 35(10): 2408–2416.
Van den Berghe, G. 2004. How does blood glucose control with insulin save lives in intensive care? Journal of Clinical Investigation 114(9): 1187–1195.
Rolo, A.P., J.S. Teodoro, and C.M. Palmeira. 2012. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radical in Biology & Medicine 52(1): 59–69.
Zhang, L.N., J.J. Zheng, L. Zhang, X. Gong, H. Huang, C.D. Wang, et al. 2011. Protective effects of asiaticoside on septic lung injury in mice. Experimental Toxicology and Pathology 63(6): 519–525.
Kaplan, J.M., M. Nowell, P. Lahni, M.P. O’Connor, P.W. Hake, and B. Zingarelli. 2012. Short-term high fat feeding increases organ injury and mortality after polymicrobial sepsis. Obesity (Silver Spring) 20(10): 1995–2002.
Steinberg, J., J. Halter, H.J. Schiller, M. Dasilva, S. Landas, L.A. Gatto, et al. 2003. Metalloproteinase inhibition reduces lung injury and improves survival after cecal ligation and puncture in rats. Journal of Surgical Research 111(2): 185–195.
Cadirci, E., Z.A. Berrin, H. Zekai, O. Fehmi, M.H. Uyanik, C. Gundogdu, et al. 2010. α-lipoic acid as a potential target for the treatment of lung injury caused by cecal ligation and puncture-induced sepsis model in rats. Shock 33(5): 479–484.
Bacanlı, M., S. Aydın, G. Taner, H.G. Göktaş, T. Şahin, A.A. Başaran, et al. 2014. The protective role of ferulic acid on sepsis-induced oxidative damage in Wistar albino rats. Environmental Toxicology and Pharmacology 38(3): 774–782.
Taner, G., S. Aydın, M. Bacanlı, Z. Sarıgöl, T. Sahin, A.A. Başaran, et al. 2014. Modulating effects of pycnogenol® on oxidative stress and DNA damage induced by sepsis in rats. Phytotherapy Research 28(11): 1692–1700.
Arvidsson, S., K. Falt, S. Marklund, and U. Haglund. 1985. Role of free oxygen radicals in the development of gastrointestinal mucosal damage in Escherichia coli sepsis. Circulatory Shock 16(4): 383–393.
Xiao, F., S. Pardue, T.Y. Aw, and D.L. Carden. 2005. Obesity exacerbates sepsis-mediated pulmonary microvascular injury. Academic Emergency Medicine 12(5 suppl 1): 37–38.
Mruk, D.D., B. Silvestrini, M.Y. Mo, and C.Y. Cheng. 2002. Antioxidant superoxide dismutase—a review: its function, regulation in the testis, and role in male fertility. Contraception 65(4): 305–311.
Glorieux, C., M. Zamocky, J.M. Sandoval, J. Verrax, and P.B. Calderon. 2015. Regulation of catalase expression in healthy and cancerous cells. Free Radical in Biology & Medicine 87: 84–97.
Acknowledgments
This research was supported by the Programa de Pós-graduação em Ciências da Saúde–UNISUL and the CNPq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All studies were performed in compliance with the National Institutes of Health Guidelines and with the approval of the Animal Care and Experimentation Committee of UNISUL (protocol number 13.026.4.03.IV), Brazil.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Petronilho, F., Giustina, A.D., Nascimento, D.Z. et al. Obesity Exacerbates Sepsis-Induced Oxidative Damage in Organs. Inflammation 39, 2062–2071 (2016). https://doi.org/10.1007/s10753-016-0444-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-016-0444-x