
Abstract
Macrophage migration inhibitory factor (MIF) plays a critical role in inflammation and is elevated in diabetic kidney. However, whether MIF plays a causative role in diabetic nephropathy (DN) remains unclear. In the present study, we have demonstrated that after treatment of 8-week-old diabetic db/db and nondiabetic db/m mice with the MIF inhibitor ISO-1 (20 mg/kg) for 8 weeks, there was a significant decrease in blood glucose, albuminuria, extracellular matrix accumulation, epithelial–mesenchymal transition (EMT), and macrophage activation in the kidney of db/db mice. Incubation of macrophages with MIF induced the production of proinflammatory cytokines, including interleukin (IL) 6, IL-1β, tumor necrosis factor α (TNF-α). The conditioned media (CM) of MIF-activated macrophages and TNF-α induced by MIF caused podocyte damage. Moreover, CM from MIF-activated macrophages induced EMT of renal tubular cells, and this effect was blocked by ISO-1. Thus, MIF inhibition may be a potential therapeutic strategy for DN. This effect may be attributable to its inhibitory effect on macrophage activation in the diabetic kidney.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Diabetic nephropathy (DN) is the leading cause of chronic kidney disease and is one of the most significant long-term complications in terms of morbidity and mortality for patients with diabetes. Although DN is traditionally considered a nonimmune disease, accumulating evidence indicates that immunologic and inflammatory mechanisms play a significant role in its development and progression [24, 36]. Infiltration of inflammatory cells and secretion of inflammatory cytokines and chemokines from the inflammatory cells have been shown to be associated with DN [11, 14, 27, 30]. Indeed, anti-inflammation is considered as a potential therapeutic approach for DN [16].
Macrophage migration inhibitory factor (MIF), a widely expressed pleiotropic cytokine, exhibits a broad range of immune and inflammatory activities. Previous studies have recognized MIF as the first molecule to arrive at the inflammation site and the factor that likely determines the degree of cellular inflammation [21]. MIF is known to counter-regulate the immunosuppressive effects of steroids [7] and to over-regulate the expression of Toll-like receptor (TLR)-4 on antigen-presenting cells [29]. MIF has been implicated in both types of diabetes [32], and there is evidence linking MIF with DN. MIF is elevated in the serum of individuals with type 2 diabetes [13] and is also increased in experimental DN [6], preceding the onset of microalbuminuria [37]. In addition, the MIF receptor, CD74, is also upregulated in clinical and experimental DN [31]. Based on the finding that MIF transgenic mice develop progressive glomerulosclerosis with proteinuria and renal failure [33], it is hypothesized that increased MIF expression may contribute DN pathogenesis.
In the present study, we investigated the renal effects of MIF inhibition in a diabetic experimental model. Inhibition of MIF tautomerase activity with the small molecular inhibitor ISO-1 ((S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester) has been shown to significantly inhibit MIF’s proinflammatory activities [1]. We observed that treatment of the diabetic db/db mice with the MIF inhibitor ISO-1 significantly decreased blood glucose level, albuminuria, extracellular matrix accumulation, and epithelial–mesenchymal transition (EMT) in the kidney. These effects may be attributable to its inhibitory effect on macrophage activation in the diabetic kidney. Thus, MIF inhibition may be a potential therapeutic strategy for DN.
MATERIALS AND METHODS
Animals
Six-week-old C57BLKS/J db/db and db/m male mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice had free access to tap water and were fed with a standard chow diet. Animals were kept under 12-h light/dark cycles.
ISO-1 Treatment
MIF inhibition was achieved by ISO-1 (Merck Chemicals, Darmstadt, Germany), a widely used pharmacological compound that binds to the tautomerase active site of MIF [2, 20]. Mice received ISO-1 intraperitoneal injections (20 mg/kg total body weight) twice a week or the equivalent amount of DMSO used as solvent [22] for 8 weeks, starting at 8 weeks of age. All experiments were conducted in accordance with the National Institutes of Health guidelines and with the approval of the University Institutional Animal Care and Use Committee.
Urine Collection and Measurement of Urinary Albumin Excretion
Mice were placed individually in metabolic cages for 24-h urine collection with free access to food and water. Urine specimens were collected every 12 h and kept at 4 °C until the 24-h collection period was completed. To prevent protein degradation, a total of 10 μl of protease inhibitor (Roche Diagnostics, IN, USA) was added to the collection tubes during the 24-h collection period. Urine samples were centrifuged at 10,000 × g for 5 min at 4 °C to remove debris, and supernatants were aliquoted and stored at −80 °C for later use. To measure urinary albumin excretion, individual mice were placed once every month in a metabolic cage and urine was collected for 18 h. Urinary albumin concentrations were determined by enzyme-linked immunosorbent assay (ELISA; Exocell, Philadelphia, PA) after the correction by urinary creatinine concentrations.
Blood Glucose Levels and Glucose Tolerance Test
Whole blood glucose levels on a drop of tail blood collected between 10 a.m. and midday, or at intervals thereafter as indicated were measured using FreeStyle® Blood Glucose Test Strips and FreeStyle Lite® Blood Glucose Monitoring System (Abbott, CA, USA). For glucose tolerance test, mice were fasted overnight for 16 h and injected with glucose (1.5 mg/kg, intraperitoneally (I.P.)). Blood samples (5 μl) were collected in heparinized capillary tubes from the tip of the tail at 0, 15, 30, 45, 60, 90, and 120 min after glucose injection and diluted in 10 % Lysis M Reagent (Roche Diagnostics, IN, USA). Glucose concentration was determined colorimetrically using a glucose oxidase/peroxidase reagent kit (Sigma, MO, USA). Samples were incubated with 100 μl of assay reagent at 37 °C for 30 min. The reaction was stopped by adding 12 N H2SO4 and measured at 540 nm using a Fusion Packard plate reader. Values were expressed in milligrams per deciliter. Area under the glucose tolerance curve (0–120 min) was carried out using Prism software.
Preparation of Bone Marrow-Derived Macrophages
Macrophages were generated from mouse bone marrow cells. The protocols for animal handling were previously approved by our Institutional Animal Care and Use Committee. To prepare murine bone marrow cells, femurs were obtained from 6- to 12-week-old C57BL/6 mice. After euthanasia, the mice were sprayed with 70 % ethanol and the femurs were dissected using scissors, cutting through the tibia below the knee joints, as well as through the pelvic bone close to the hip joint. Muscles connected to the bone were removed using clean gauze, and the femurs were placed into a polypropylene tube containing sterile phosphate-buffered saline (PBS) on ice. In a tissue culture hood, the bones were placed in 70 % ethanol for 1 min, washed in sterile RPMI 1640, and then both epiphyses were removed using sterile scissors and forceps. The bones were flushed with a syringe filled with RPMI 1640 to extrude bone marrow into a 15-ml sterile polypropylene tube. A 5-ml plastic pipette was used to gently homogenize the bone marrow. The cell suspension generated thereafter is called fresh bone marrow cells.
Bone marrow-derived macrophages (BMDM) were prepared as previously described [41] using L929-cell conditioned medium (LCCM) as a source of granulocyte/macrophage colony-stimulating factor [10]. The cells were resuspended in 10-ml bone marrow differentiation media (R20/30), which is RPMI 1640 supplemented with 20 % fetal bovine serum (Gibco, cat. 12657-029), 30 % LCCM, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. Cells were seeded in nontissue culture-treated Optilux Petri dishes (BD Biosciences) and incubated at 37 °C in a 5 % CO2 atmosphere. Four days after seeding the cells, an extra 10 ml of fresh R20/30 were added per plate and incubated for an additional 3 days. To obtain the BMDM, the supernatants were discarded and the attached cells were washed with 10 ml of sterile PBS. Ten milliliters of ice-cold PBS were added to each plate and incubated at 4 °C for 10 min. The macrophages were detached by gently pipetting the PBS across the dish. The cells were centrifuged at 200 × g for 5 min and resuspended in 10 ml of BMDM cultivation media (R10/5), which is composed of RPMI 1640, 10 % fetal bovine serum, 5 % LCCM, and 2 mM L-glutamine. The cells were counted, seeded, and cultivated in tissue culture plates 12 h before any further experimental procedure.
Mouse Podocyte Cell Culture
A mouse podocyte cell line was donated by Dr. Peter Mundel (Albert Einstein College of Medicine, Bronx, NY) [12] Cultivation of conditionally immortalized mouse podocytes was conducted as reported previously [26]. In brief, podocytes between passage 14 and 20 were maintained in RPMI 1640 medium supplemented with 10 % FBS. Cells were propagated in 10 U/ml murine interferon gamma (IFN-γ) at 33 °C, and then differentiated by culture for 1 week at 37 °C in the absence of IFN-γ. Differentiated cells showed prominent cytoplasmic processes and expressed nephrin and podocin as shown previously [26].
Mouse Renal Tubular Cell Culture
A mouse renal tubular cell line (MCT) was kindly provided by Dr. Neilson (University of Pennsylvania) [39]. Cells were maintained in Dulbecco's modified Eagles's medium (DMEM; GIBCO) and 10 % fetal calf serum (FCS) as previously reported [28].
ELISA
Tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, and MIF were measured in protein extracts from whole kidney cortex. Fifty milligrams of kidney cortex was homogenized in PBS and 0.5 mm zirconiumoxide beads for 4 min using a bullet blender system (Next Advance, Averill Park, NY, USA). The MIF, TNF-α, and IL-6 ELISA were performed according to the manufacturer’s instructions (Quantikine, R&D Systems, Minneapolis, MN, USA).
Western Blotting
The kidney tissues or cells were homogenized in RIPA buffer and spun at 14, 000 × g to pellet the nuclei and large cellular fragments. The supernatant protein concentrations were measured by the BCA assay (Bio-Rad, Hercules) and equalized with the addition of Laemmli buffer, before sodium dodecyl sulfate (SDS)-based electrophoresis through a 6–10 % SDS polyacrylamide gel. After electrical wet transfer of the proteins to a nitrocellulose membrane, the membranes were incubated with primary antibodies at 4 °C overnight. After incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies, the chemiluminescent reaction was developed with SuperSignal West Pico (Pierce, Rockford, IL). The following primary antibodies were used: mouse anti-CD86 (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-inducible nitric oxide synthase (iNOS; 1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-nephrin (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-podocin (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-fibronectin (1:1,000, Abcam), mouse anti-collagen I (1:1,000, Abcam), rabbit anti-α-SMA (1:2,000, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-E-cadherin (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), and goat anti-β-actin (1:5,000, Abcam).
RNA Preparation and Quantitative Real-Time PCR
RNA was extracted using a standard TRIzol-based protocol, according to manufacturer’s instructions. Known concentrations of purified RNA were used for complementary DNA (cDNA) synthesis. RNA was reverse transcribed using SuperScript II and random primers (Invitrogen) to obtain cDNA. Quantitative real-time PCR analysis was conducted by using specific primers for TNF-a, IL-1b, IL-6, and iNOS, as previously described [9, 17]. Briefly, PCRs were carried out using 10 ng cDNA, 500 nM forward and reverse primers, and SYBR green master mix (Applied Biosystems) in 20 ml reactions. Each sample was analyzed in triplicate, and SYBR green fluorescence was detected using the Applied Biosystems 7900HT real-time PCR system. A standard curve was generated for each gene by using a cDNA dilution series, and melting point analysis was performed to confirm a single amplification product. Data were normalized to the 18S gene, and expressed as fold change from control.
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). Differences between the groups were examined for statistical significance using one-way ANOVA with the Tukey’s correction (SPSS 11.5; SPSS, Chicago, IL) for multiple comparisons when appropriate. A p value of <0.05 was considered to be a statistically significant difference.
RESULTS
ISO-1 Normalized Hyperglycemia and Improved Glucose Metabolism in db/db Mice
Eight-week-old db/db mice exhibited significantly higher blood glucose levels, which consistently increased throughout the study periods, compared with control mice (Fig. 1a). Chronic treatment of db/db mice with ISO-1 (20 mg/kg) significantly decreased blood glucose after 1 week and normalized hyperglycemia throughout the 8-week duration of treatment (Fig. 1a). Using an intraperitoneal glucose tolerance test, db/db mice showed impaired glucose utilization compared to age-matched control mice, which was significantly improved in ISO-1-treated db/db mice (Fig. 1b, c). These effects were not observed in lean control mice treated with ISO-1.

Chronic treatment with ISO-1 (20 mg/kg) normalized hyperglycemia and improved glucose tolerance in db/db mice. a Nonfasted blood glucose levels in db/m, db/m + ISO-1, db/db, and db/db + ISO-1 mice. Data are represented as mean ± SEM of each group (n = 7–9). b Glucose tolerance test in ISO-1-treated and untreated lean control and db/db mice. After 8 weeks of treatment with ISO-1, mice were fasted for 16 h and dosed with glucose (1.5 g/kg, I.P.). Blood glucose levels were measured by tail tip bleed at 0, 15, 30, 60, 90, and 120 min postadministration. Data are represented as mean ± SEM of each group (n = 7–9). c One-way ANOVA of area under curve showed that ISO-1 significantly improved the glucose tolerance in db/db + ISO-1 mice compared with untreated db/db mice. *p < 0.05 vs. age-matched lean control mice; # p < 0.05 vs. untreated db/db mice.
ISO-1 Treatment Ameliorated Diabetic Albuminuria
To assess the effect of ISO-1 treatment on albuminuria, we measured the total urinary albumin excretion rate and the urinary albumin-to-creatinine ratio in the 18-h urine collections. The albumin excretion rate was markedly increased in the diabetic db/db mice compared with the nondiabetic db/m control mice. Interestingly, urinary albumin excretion was significantly ameliorated after ISO-1 treatment (Fig. 2).

Chronic treatment with ISO-1 reduced albuminuria in db/db mice. Total urinary albumin excretion (UAE) or urinary albumin creatinine ratio (UAC) were significantly attenuated in diabetic mice treated with ISO-1. Data are represented as mean ± SEM of each group (n = 7–9). # p < 0.05 vs. untreated nondiabetic mice; *p < 0.05 vs. untreated diabetic mice.
ISO-1 Treatment Prevented Renal Macrophage Infiltration in Diabetes
The effect of ISO-1 on renal macrophage accumulation was evaluated by Western blotting. The amount of CD86 and iNOS, corrected for β-actin, in the kidney tissues was found to be increased in the diabetic mice compared with nondiabetic controls (Fig. 3a, b). The increase in renal CD86 and iNOS with diabetes was significantly diminished by ISO-1 treatment (Fig. 3a, b). Inflammatory markers in kidney tissue, i.e., IL-6 (Fig. 3c) and TNF-α (Fig. 3d), were also measured. Both IL-6 and TNF-α were markedly increased in diabetic mice compared with control mice. Treatment of the diabetic mice with ISO-1 significantly decreased the levels of IL-6 and TNF-α as compared with the untreated diabetic mice (Fig. 3c, d). Renal MIF levels were significantly increased in diabetic mice (2.0-fold vs. nondiabetic control; Fig. 3e). ISO-1 treatment did not significantly change renal MIF levels in the diabetic mice (Fig. 3e).

Chronic treatment with ISO-1 reduced renal macrophage activation and production of inflammatory cytokines. a Representative Western blot images showed a marked increase in renal CD86 and iNOS expression in the diabetic mice compared with nondiabetic controls, which was diminished by ISO-1 treatment. b Quantification of the amount of CD86 and iNOS evaluated by Western blotting, corrected for β-actin, indicated a significant increase in the diabetic mice compared with nondiabetic controls. The increase in renal CD86 and iNOS with diabetes was significantly diminished by ISO-1 treatment. Data are represented as mean ± SEM of each group (n = 7–9). # p < 0.05 vs. untreated nondiabetic mice with ISO-1; *p < 0.05 vs. untreated diabetic mice. c, d Levels of IL-6 and TNF-α in kidney were measured by ELISA. Data are represented as mean ± SEM of each group (n = 7–9). # p < 0.05 vs. untreated nondiabetic mice; *p < 0.05 vs. untreated diabetic mice. e Renal levels of MIF were measured by ELISA. Data are represented as mean ± SEM of each group (n = 7–9). *p < 0.05 compared with nondiabetic control.
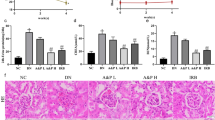
ISO-1 Treatment Reduced ECM Expression and EMT in the Kidneys of db/db Mice
We examined whether ISO-1 treatment can prevent ECM accumulation and EMT in the kidneys of db/db mice. Glomerular (Fig. 4a) and tubular (Fig. 4b) fibronectin and collagen I expression were significantly increased, whereas tubular E-cadherin expression was significantly decreased in the db/db mice compared with control mice. ISO-1 effectively prevented the increases in fibronectin and collagen I and the decrease in E-cadherin expression levels in db/db mice, whereas it had no effect in control mice (Fig. 4).

Chronic treatment with ISO-1 reduced extracellular matrix (ECM) expression and epithelial-to-mesenchymal transition (EMT) in the kidneys of db/db mice. After the treatment of db/db mice with ISO-1 (20 mg/kg) for 8 weeks, glomerular expression of fibronectin and collagen I (a) and tubular expression of fibronectin, collagen I, and E-cadherin (b) were analyzed by Western blotting. Data are means ± SEM (n = 7–9 per group). # p < 0.05 vs. untreated nondiabetic mice 1; *p < 0.05 vs. untreated db/db mice.
CM from MIF-Activated Macrophages Downregulated Nephrin and Podocin Expressions
Incubation of macrophages with MIF resulted in increased expression of iNOS and a number of proinflammatory cytokines, including TNF-α, IL-6, and IL-1β (Fig. 5a). To examine whether MIF or the inflammatory cytokines from MIF-activated macrophages plays a role in podocyte damage, nephrin and podocin expression levels were measured with Western blotting after MIF treatment. Incubation of differentiated mouse podocytes with TNF-α significantly reduced the expression levels of nephrin and podocin, consistent with a previous report [15]. However, we did not observe a significant effect of MIF, IL-6, or IL1β on the expression of nephrin or podocin (Fig. 5b, c). We also examined whether CM from MIF-activated macrophages causes podocyte damage. Incubation of differentiated mouse podocytes with CM from unstimulated macrophages had no effect upon nephrin or podocin expression (data not shown). By contrast, CM from MIF-activated macrophages caused a significant reduction in both nephrin and podocin expressions (Fig. 5c). This effect was blocked by a TNF-α neutralizing antibody (Fig. 5c).

CM and TNF-α in the CM from MIF-activated macrophages reduced nephrin and podocin expression in podocyte cultures. a After the incubation of bone marrow-derived macrophages with MIF (0, 1, or 10 nM) for 6 h, the relative gene expression of TNF-α, IL-1β, IL-6, and iNOS was determined by RT-PCR. Bar graph shows mean ± SEM of each group. *p < 0.05; **p < 0.01 vs. vehicle treatment. b Podocyte cultures were incubated with MIF (10 nM), TNF-α (10 nM), or IL-6 (10 nM) for 24 h, and the expression of nephrin and podocin was assessed with Western blotting. Quantification of the amount of nephrin and podocin immunoblots, corrected for β-actin immunoblots, indicated a significant decrease in nephrin and podocin expression in TNF-α-treated cells. Data are mean ± SEM from three independent experiments. *p < 0.05 vs. vehicle treatment. c Podocyte cultures were incubated with CM from unstimulated (CM-none) or MIF-stimulated macrophages (MIF-CM), with or without a neutralizing anti-TNF-a antibody for 24 h, and the expression of nephrin and podocin was assessed with Western blotting. Quantification of the amount of nephrin and podocin immunoblots, corrected for β-actin immunoblots, indicated a significant decrease in nephrin and podocin expression in MIF-CM-treated cells, which was diminished by anti-TNF-α antibody. Data are mean ± SEM from three independent experiments. # p < 0.05 vs. CM-none treatment; *p < 0.05 vs. MIF-CM treatment.
CM from MIF-Activated Macrophages Enhanced ECM Expression and EMT of MCT
Incubation of MCT with either MIF or CM from unstimulated macrophages had no effect on the expression of fibronectin, collagen I, α-SMA, or E-cadherin (data not shown). By contrast, incubation of MCT with CM from MIF-activated macrophages significantly increased fibronectin, collagen I, and α-SMA expression levels and significantly decreased E-cadherin expression compared with the control CM from unstimulated macrophages. Intriguingly, treatment of the CM from both MIF and ISO-1-treated macrophages did not affect the expression of fibronectin, collagen I, α-SMA, or E-cadherin in the MCT cells (Fig. 6). These data suggest that MIF induces the production of fibrogenic factors from macrophages, and this process can be blocked by ISO-1.

CM from MIF-activated macrophages induced EMT of renal tubular cells, which was diminished by ISO-1. Murine renal tubular cell line culture was incubated with MIF, CM from unstimulated (CM-none) or CM from MIF-stimulated macrophages (MIF-CM) pretreated with or without ISO-1 for 24 h, and the expression levels of fibronectin, collagen I, α-SMA, and E-cadherin were assessed with Western blotting. Representative Western blots (a) and quantitative data (mean ± SEM) of the amount of fibronectin, collagen I, α-SMA, and E-cadherin from three independent experiments, corrected for β-actin immunoblots (b), were shown. # p < 0.05 vs. CM-none treatment; *p < 0.05 vs. MIF-CM treatment.
DISCUSSION
In the present study, we demonstrate for the first time that ISO-1 normalized hyperglycemia and improves glucose handling, reduced albuminuria, and prevented ECM accumulation and EMT in the kidney of db/db mice. ISO-1 treatment significantly reduced macrophage activation, associated with marked decrease in ECM accumulation and EMT, in the diabetic kidneys. In the podocyte culture, MIF did not directly induce podocyte damage, but CM from MIF-activated macrophages and TNF-α induced by MIF in the macrophage culture did. Moreover, CM from MIF-activated macrophages induced EMT of renal tubular cells, which was abolished by ISO-1. These findings suggest that MIF inhibition may be beneficial to DN patients and this effect may be attributable to its modulation of macrophage activation.
Like most other forms of chronic kidney diseases, DN is characterized by progressive fibrosis as a final pathway that eventually affects all substructures of the kidney. Recent studies have revealed major pathways leading to the development of renal interstitial fibrosis following unilateral ureteral obstruction: (1) interstitial infiltration of macrophages that produce cytokines responsible for tubular apoptosis and fibroblast proliferation and activation; (2) tubular cell death leading to the formation of tubular atrophy; and (3) phenotypic transition of resident renal cells. Chronic inflammation, attributed largely from aberrant macrophage activation, is considered as a major player in this pathogenic process. Recent studies have demonstrated that classically activated M1 macrophages, but not M2 macrophages, induced podocyte permeability [40]. It is also proved by other studies that M1 macrophages can generate TNF-α, which mediates proapoptotic signaling and renal tubular cell apoptosis following unilateral ureteral obstruction [23], whereas M2 macrophages generate anti-inflammatory cytokines and trophic factors that induce cell survival and proliferation [8].
We are interested in MIF as a potential therapeutic target for DN not only because it is elevated in DN patients and animal models [6, 13, 35, 37] but also due to its pivotal role in inflammation cascade and macrophage polarization. MIF modulates the production or expression of several proinflammatory molecules, including cytokines, nitric oxide, and in the activation of the cyclooxygenase pathway [3], thereby playing an important role in the pathophysiology of several inflammatory diseases [3]. More importantly, recent studies have pointed out an important role of MIF in macrophage accumulation and polarization. For instance, MIF deficiency results in reduced expression of inflammatory mediators (e.g., IL-1β, CCL2, and MMPs) and increased production of anti-inflammatory cytokines (e.g., IL-10) in the lesion area [38]. These findings, together with our data that incubation of macrophage cultures with MIF resulted in increased expression of TNF-α, IL-6, IL-1β, and iNOS, all of which are M1 macrophage markers, suggest an important role of MIF in macrophage polarization to M1.We provided evidence that the modulation of macrophage activation and polarization by MIF is of functional significance: (1) the CM from MIF-activated macrophages caused podocyte damage and EMT of renal tubular cells in vitro, and (2) treatment of db/db mice with ISO-1 resulted in reduced macrophage activation in the diabetic kidney, associated with reduced blood glucose level, albuminuria, ECM accumulation and EMT in vivo.
As shown in the present study, whereas TNF-α produced by MIF-activated macrophages contributed to podocyte damage, it did not directly induce EMT of renal tubular cells, suggesting that different mechanisms are involved in podocyte damage and fibrosis. Though mechanisms underlying the fibrogenic role of MIF-activated macrophages in the diabetic kidney remain to be investigated, previous studies have shown that MIF induces the expression of several fibrosis mediators, including but not limited to TGF-β1, ICAM-1, and MMPs [5, 19, 34], all of which are able to induce EMT of renal tubular cells [4, 18, 25].
Altogether, we demonstrated that treatment of the diabetic db/db mice with the MIF inhibitor ISO-1 significantly decreased blood glucose level, albuminuria, ECM accumulation, and EMT in the diabetic kidney. These effects may be attributable to its inhibitory effect on macrophage activation in the diabetic kidney. Thus, MIF inhibition may be a potential therapeutic strategy for DN.
References
Al-Abed, Y., D. Dabideen, B. Aljabari, A. Valster, D. Messmer, M. Ochani, M. Tanovic, K. Ochani, M. Bacher, F. Nicoletti, C. Metz, V.A. Pavlov, E.J. Miller, and K.J. Tracey. 2005. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. The Journal of Biological Chemistry 280(44): 36541–36544.
Al-Abed, Y., and S. VanPatten. 2011. MIF as a disease target: ISO-1 as a proof-of-concept therapeutic. Future Medicinal Chemistry 3(1): 45–63.
Calandra, T., and T. Roger. 2003. Macrophage migration inhibitory factor: A regulator of innate immunity. Nature Reviews Immunology 3(10): 791–800.
Catania, J.M., G. Chen, and A.R. Parrish. 2007. Role of matrix metalloproteinases in renal pathophysiologies. American Journal of Physiology. Renal Physiology 292(3): F905–F911.
Cheng, Q., S.J. McKeown, L. Santos, F.S. Santiago, L.M. Khachigian, E.F. Morand, and M.J. Hickey. 2010. Macrophage migration inhibitory factor increases leukocyte–endothelial interactions in human endothelial cells via promotion of expression of adhesion molecules. Journal of Immunology 185(2): 1238–1247.
Chow, F., E. Ozols, D.J. Nikolic-Paterson, R.C. Atkins, and G.H. Tesch. 2004. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney International 65(1): 116–128.
de Jong, Y.P., A.C. Abadia-Molina, A.R. Satoskar, K. Clarke, S.T. Rietdijk, W.A. Faubion, E. Mizoguchi, C.N. Metz, M. Alsahli, T. ten Hove, A.C. Keates, J.B. Lubetsky, R.J. Farrell, P. Michetti, S.J. van Deventer, E. Lolis, J.R. David, A.K. Bhan, and C. Terhorst. 2001. Development of chronic colitis is dependent on the cytokine MIF. Nature Immunology 2(11): 1061–1066.
Duffield, J.S. 2003. The inflammatory macrophage: A story of Jekyll and Hyde. Clinical Science 104(1): 27–38.
Ejarque-Ortiz, A., M.G. Medina, J.M. Tusell, A.P. Perez-Gonzalez, J. Serratosa, and J. Saura. 2007. Upregulation of CCAAT/enhancer binding protein beta in activated astrocytes and microglia. Glia 55(2): 178–188.
Englen, M.D., Y.E. Valdez, N.M. Lehnert, and B.E. Lehnert. 1995. Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. Journal of Immunological Methods 184(2): 281–283.
Furuta, T., T. Saito, T. Ootaka, J. Soma, K. Obara, K. Abe, and K. Yoshinaga. 1993. The role of macrophages in diabetic glomerulosclerosis. American Journal of Kidney Diseases: The Official Journal of the National Kidney Foundation 21(5): 480–485.
Greka, A., and P. Mundel. 2012. Cell biology and pathology of podocytes. Annual Review of Physiology 74: 299–323.
Herder, C., H. Kolb, W. Koenig, B. Haastert, S. Muller-Scholze, W. Rathmann, R. Holle, B. Thorand, and H.E. Wichmann. 2006. Association of systemic concentrations of macrophage migration inhibitory factor with impaired glucose tolerance and type 2 diabetes: Results from the Cooperative Health Research in the Region of Augsburg, Survey 4 (KORA S4). Diabetes Care 29(2): 368–371.
Hirata, K., K. Shikata, M. Matsuda, K. Akiyama, H. Sugimoto, M. Kushiro, and H. Makino. 1998. Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia 41(2): 185–192.
Ikezumi, Y., T. Suzuki, T. Karasawa, H. Kawachi, D.J. Nikolic-Paterson, and M. Uchiyama. 2008. Activated macrophages down-regulate podocyte nephrin and podocin expression via stress-activated protein kinases. Biochemical and Biophysical Research Communications 376(4): 706–711.
Impellizzeri, D., E. Esposito, J. Attley, and S. Cuzzocrea. 2014. Targeting inflammation: New therapeutic approaches in chronic kidney disease (CKD). Pharmacological Research: The Official Journal of the Italian Pharmacological Society 81C: 91–102.
Kigerl, K.A., W. Lai, S. Rivest, R.P. Hart, A.R. Satoskar, and P.G. Popovich. 2007. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. Journal of Neurochemistry 102(1): 37–50.
Lan, H.Y., and A.C. Chung. 2012. TGF-beta/Smad signaling in kidney disease. Seminars in Nephrology 32(3): 236–243.
Leung, J.C., L.Y. Chan, A.W. Tsang, E.W. Liu, M.F. Lam, S.C. Tang, and K.N. Lai. 2004. Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association—European Renal Association 19(8): 1976–1985.
Lubetsky, J.B., A. Dios, J. Han, B. Aljabari, B. Ruzsicska, R. Mitchell, E. Lolis, and Y. Al-Abed. 2002. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. The Journal of Biological Chemistry 277(28): 24976–24982.
Malorny, U., M. Goebeler, J. Gutwald, J. Roth, and C. Sorg. 1990. Differences in migration inhibitory factor production by C57Bl/6 and BALB/c mice in allergic and irritant contact dermatitis. International Archives of Allergy and Applied Immunology 92(4): 356–360.
Meyer-Siegler, K.L., K.A. Iczkowski, L. Leng, R. Bucala, and P.L. Vera. 2006. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. Journal of Immunology 177(12): 8730–8739.
Misseri, R., D.R. Meldrum, C.A. Dinarello, P. Dagher, K.L. Hile, R.C. Rink, and K.K. Meldrum. 2005. TNF-alpha mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. American Journal of Physiology. Renal Physiology 288(2): F406–F411.
Mora, C., and J.F. Navarro. 2006. Inflammation and diabetic nephropathy. Current Diabetes Reports 6(6): 463–468.
Morishita, Y., M. Watanabe, E. Nakazawa, K. Ishibashi, and E. Kusano. 2011. The interaction of LFA-1 on mononuclear cells and ICAM-1 on tubular epithelial cells accelerates TGF-beta1-induced renal epithelial–mesenchymal transition. PLoS ONE 6(8): e23267.
Mundel, P., J. Reiser, A. Zuniga Mejia Borja, H. Pavenstadt, G.R. Davidson, W. Kriz, and R. Zeller. 1997. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Experimental Cell Research 236(1): 248–258.
Navarro-Gonzalez, J.F., and C. Mora-Fernandez. 2008. The role of inflammatory cytokines in diabetic nephropathy. Journal of the American Society of Nephrology: JASN 19(3): 433–442.
Okada, H., T.M. Danoff, R. Kalluri, and E.G. Neilson. 1997. Early role of Fsp1 in epithelial–mesenchymal transformation. The American Journal of Physiology 273(4 Pt 2): F563–F574.
Roger, T., C. Froidevaux, C. Martin, and T. Calandra. 2003. Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). Journal of Endotoxin Research 9(2): 119–123.
Ruster, C., and G. Wolf. 2008. The role of chemokines and chemokine receptors in diabetic nephropathy. Frontiers in Bioscience: A Journal and Virtual Library 13: 944–955.
Sanchez-Nino, M.D., A.B.. Sanz, P. Ihalmo, M. Lassila, H. Holthofer, S. Mezzano, C. Aros, P.H. Groop, M.A. Saleem, P.W. Mathieson, R. Langham, M. Kretzler, V. Nair, K.V. Lemley, R.G. Nelson, E. Mervaala, D. Mattinzoli, M.P. Rastaldi, M. Ruiz-Ortega, J.L. Martin-Ventura, J. Egido, and A. Ortiz. 2009. The MIF receptor CD74 in diabetic podocyte injury. Journal of the American Society of Nephrology: JASN 20(2): 353–362.
Sanchez-Zamora, Y.I., and M. Rodriguez-Sosa. 2014. The role of MIF in type 1 and type 2 diabetes mellitus. Journal of Diabetes Research 2014: 804519.
Sasaki, S., J. Nishihira, T. Ishibashi, Y. Yamasaki, K. Obikane, M. Echigoya, Y. Sado, Y. Ninomiya, and K. Kobayashi. 2004. Transgene of MIF induces podocyte injury and progressive mesangial sclerosis in the mouse kidney. Kidney International 65(2): 469–481.
Shyu, L.Y., T.M. Yeh, H.H. Chang, D.P. Lin, Y.H. Teng, L.C. Chen, and H.H. Lee. 2012. Macrophage migration inhibitory factor induces ICAM-1and thrombomobulin expression in vitro. Thrombosis Research 129(1): 43–49.
Tashimo, A., Y. Mitamura, S. Nagai, Y. Nakamura, K. Ohtsuka, Y. Mizue, and J. Nishihira. 2004. Aqueous levels of macrophage migration inhibitory factor and monocyte chemotactic protein-1 in patients with diabetic retinopathy. Diabetic Medicine: A Journal of the British Diabetic Association 21(12): 1292–1297.
Tuttle, K.R. 2005. Linking metabolism and immunology: Diabetic nephropathy is an inflammatory disease. Journal of the American Society of Nephrology: JASN 16(6): 1537–1538.
Watanabe, T., N.H. Tomioka, M. Doshi, S. Watanabe, M. Tsuchiya, and M. Hosoyamada. 2013. Macrophage migration inhibitory factor is a possible candidate for the induction of microalbuminuria in diabetic db/db mice. Biological & Pharmaceutical Bulletin 36(5): 741–747.
White, D.A., Y. Su, P. Kanellakis, H. Kiriazis, E.F. Morand, R. Bucala, A.M. Dart, X.M. Gao, and X.J. Du. 2014. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. Journal of Molecular and Cellular Cardiology 69: 32–42.
Wolf, G., G.S. Kuncio, M.J. Sun, and E.G. Neilson. 1991. Expression of homeobox genes in a proximal tubular cell line derived from adult mice. Kidney International 39(5): 1027–1033.
You, H., T. Gao, T.K. Cooper, W. Brian Reeves, and A.S. Awad. 2013. Macrophages directly mediate diabetic renal injury. American Journal of Physiology. Renal Physiology 305(12): F1719–F1727.
Zamboni, D.S., and M. Rabinovitch. 2003. Nitric oxide partially controls Coxiella burnetii phase II infection in mouse primary macrophages. Infection and Immunity 71(3): 1225–1233.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhigang Wang, Meng Wei, Meng Wang, Lei Chen, Hua Liu, Yi Ren, and Kehui Shi contributed equally to the work.
Rights and permissions
About this article

Cite this article
Wang, Z., Wei, M., Wang, M. et al. Inhibition of Macrophage Migration Inhibitory Factor Reduces Diabetic Nephropathy in Type II Diabetes Mice. Inflammation 37, 2020–2029 (2014). https://doi.org/10.1007/s10753-014-9934-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-014-9934-x