
Abstract
Pulmonary arterial hypertension (PAH) is a progressive and a life-threatening disease with its high morbidity and mortality ratios. On searching for new shining targets in pathogenesis, we noticed, in our previous studies, urotensin-II (UII) in systemic sclerosis with potent angiogenic and pro-fibrotic features. Owing to the mimicking properties of UII with endothelin-1 (ET1), we attempted to investigate the effect of palosuran in a PAH rat model. Thirty rats were randomly divided into three groups, with each group comprising 10 rats: group 1 (control group) received the vehicle subcutaneously, instead of monocrotaline (MCT) and vehicle; group 2 (MCT group) received subcutaneous MCT and vehicle; and group 3 (MCT + palosuran group) received subcutaneous MCT and palosuran. Serum UII, ET1, transforming growth factor-β1 (TGF-β1) levels, pulmonary arteriolar pathology of different diameter vessels, and cardiac indices were evaluated. The ET1, TGF-β1, and UII levels were significantly diminished in the treatment group, similar to the controls (p < 0.001). Right ventricular hypertrophy index and mean pulmonary arterial pressure scores were also significantly reduced in the treatment group (p = 0.001). Finally, in the 50–125-μm diameter arterioles, in contrast to Groups 3 and 1, there was a statistically significant thickness (p < 0.01) in the arteriolar walls of rats in Group 2. The treatment effect on arteries of more than 125-μm diameters was found to be valuable but not significant. Owing to its healing effect on hemodynamic, histological, and biochemical parameters of MCT-induced PAH, palosuran as an antagonist of UII might be an optional treatment alternative for PAH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The explosion in the knowledge and developments in the treatment of pulmonary arterial hypertension (PAH) has simultaneously resulted in increasing concern on this disease in the last decade. However, we are far from the satisfactory expectations regarding the mortality and morbidity ratios. The existing agents in PAH treatment are mostly targeted to increase nitric oxide by phosphodiesterase-5 inhibitors, inhibit the effects of endothelin-1 (ET1) by dual (receptor A/B) or selective (receptor A) receptor blockers, and supplement prostacyclin with prostanoids [1].
ET1 is an important peptide in vascular tonus regulation, mitogenesis, or neurohormonal balance in health status with two different receptor subtypes (A/B). However, it orchestrates vasoconstriction, fibroblast activation, vascular wall changes similar to hypertrophy, and inflammatory response, especially targeting the vessels in various diseases. The pivotal role of ET1 observed in PAH pathogenesis has resulted in initiation of a war against this cytokine and increased hope in treating primary or secondary PAH patients [2], although the results for PAH treatment, including all medications, have not been up to the expectations.
The variation in active vasculature of pulmonary arterial system in PAH could not only be explained by vasoconstriction, but also through an imbalance of the vessels with hypertrophy, angiogenesis, apoptosis, proliferation, extracellular matrix restructuring, and pro-thrombotic state. Urotensin-II (UII) is a vascular active peptide that was not only demonstrated with more potent vasoconstrictor effects than ET1 [3, 4] but also as an important proliferative, pro-thrombotic, and angiogenetic mediator of the arterial system, especially the wide-sized pulmonary vasculature [5]. The net effect of UII in normal conditions is under tight control by the downregulation of high-affinity UII receptors [6]. Thus, UII has been thought to play a major role in the pathological vascular conditions [7]. Following the idea of UII being a peptide similar to ET1 [8], we considered to explore whether UII had an effect on systemic sclerosis (SSc) pathogenesis, which is a disease with similar prominent vascular pathology as that of PAH, and demonstrated that UII levels are higher in patients with SSc, especially in diffuse fibrotic patients, in contrast to the control group. We also found a positive relationship between UII and ET1 levels [9]. Besides, in our previous study, we found an association between Thr21Met, but not Ser89Asn, in the UII gene and SSc. The results of our study strongly suggest that this single-nucleotide polymorphism may be an important risk factor for the development of SSc, and a powerful indicator of severe skin and lung involvement in patients with SSc [10].
We subsequently tried to antagonize the effects of UII in an animal PAH model to diminish the UII and ET1 levels that were presented in our preliminary studies [11]. More recently, Mei et al. demonstrated that the effect of monocrotaline (MCT)-induced PAH was antagonized with urantide, which is another UII antagonist [12].
Palosuran, an antagonist of UII, is a peptidic and orally active selective agent [13]. Earlier studies were mainly targeted to define its effectiveness, especially in renal diseases, but until now, its beneficial findings could not be obtained. In the present study, we attempt to investigate the effect of palosuran in an animal model for PAH and explore the discrepancy for pulmonary arterial pressure, UII, ET1, as well as transforming growth factor-β1 (TGF-β1) levels, and pulmonary vascular pathology.
MATERIALS AND METHODS
Animals and Reagents
In the experimental study process, 30 male Wistar albino rats (250–300 g) were purchased from Experimental Animal Center of Gaziantep University. MCT was bought from Sigma (Chemicals, St. Louis, MO), and palosuran was donated by Actelion (Gewerbestrasse 16, Allschwil CH 4123 Switzerland). The institutional committee for animal care approved all procedures involving animals.
Experimental Groups
The MCT rat model of pulmonary hypertension was used [14]. Thirty rats were randomly divided into three experimental groups, and each group included 10 rats. MCT and palosuran were dissolved in vehicle separately. MCT was administered by subcutaneous injection (60 mg/kg body weight), and palosuran was given by gavage (300 mg/kg/twice a day). The dose levels and schedules were based on previous PAH animal model studies [13, 15]. In accordance with the previous studies, the first dose of palosuran was given 2 days before the MCT injection. Palosuran and vehicle were administered by gavage twice a day from Day − 2 to Day 21 after MCT or vehicle treatment (Day 0). Group 1 (control group) received the same volume of vehicle subcutaneously, instead of MCT and vehicle by gavage. Group 2 (MCT group) received subcutaneous MCT and vehicle by gavage, while Group 3 (MCT + palosuran group) received subcutaneous MCT and palosuran by gavage. All rats were killed with exsanguination on Day 22.
Hemodynamic Study in Anesthetized Rats
To evaluate the effects of palosuran on the development of PHT, hemodynamic measurements were performed in anesthetized rats. Three weeks after MCT injection, the rats were anesthetized. A tracheotomy tube (Cwe Inc. SAR-830 Ventilator) was put in place, and a polyethylene catheter connected to a pressure transducer (Catheter: C313CT PE 50 Cannula Tubing) was inserted into the right jugular vein and passed through the right ventricle into the pulmonary artery to measure the main pulmonary arterial pressure (mPAP), in accordance with the procedure described by Stinger et al. [16]. The measurements were recorded for 15 min by using a PowerLab data acquisition system (AD Instruments Power Lab 4/25 T). The mean blood pressure (MBP) was also measured through a polyethylene catheter inserted into the left carotid artery in the same way.
Assessment of the Heart Tissue Weight Ratio
After hemodynamic measurements, chest cavity of the rats was opened. The heart was excised and placed in ice-cold normal saline to remove blood, and then was blotted and weighed. From the harvested hearts, the atria were removed, and the hearts were separated into the right and left ventricle plus septum, and weighed separately. Finally, right ventricular hypertrophy index (RVHI) was calculated as the weight ratio of right ventricle to left ventricle plus septum, and right ventricular mass index (RVMI) was calculated as the weight ratio of right ventricle to body weight [17, 18].
Morphometric Analysis of Small Pulmonary Arteries
The left lower part of the lung tissue was removed and immersed into 10 % neutral paraformaldehyde. The specimens were then dehydrated and embedded in paraffin, and sectioned at a thickness of 5 μm. The tissue incisions were stained and analyzed under light microscopy by using Hematoxylin-Eosin, Verhoeff’s Elastic stains methods. To determine and compare the increment of connective tissue and muscular structures in the lung vasculature, 30 different 50–125-μm diameters of arterioles and five different arteries with 125-μm diameter were determined at × 200 magnifications.
The wall thickness of the arteries was measured as the distance between the external and internal elastic lamina of each vessel, using a calibrated ocular micrometer. External diameter was calculated as the diameter of the external lamina. Additionally, periarterial connective tissues were measured as the distance between the external elastic lamina and lung parenchyma in 125-μm-diameter arteries using ocular micrometer, and the results were compared for statistical significance. The vessels that were not close to round or oval in shape were not measured.
ET1, UT-II, and TGFβ1 Concentrations
At the end of the hemodynamic experiments in anesthetized rats, plasma samples in EDTA were collected for determination of ET1, UII, and TGF-β1 concentrations. The plasma ET1 concentration was measured by using a human ET1 immunoassay kit (Catalog number BBE5, SBBE5, PBBE5. R&D Systems, Minneapolis, MN). UII was measured by using a rat UII enzyme immunoassay (Protocol for catalog number EK-071-09, PHOENIX PHARMACEUTICALS, INC.), and TGF-β1 was measured by using a rat TGF-β1 enzyme immunoassay (Catalog number MB100B, SMB100B, PMB100B; Quantikine).
Statistical Analysis
In the statistical evaluation of the results, the values and scores are indicated as mean values ± standard deviation. The scores and values with p < 0.05 were accepted as statistically meaningful. For the comparison of scores for more than two groups, parametric one-way ANOVA analysis was performed, and for the comparison of two groups’ meaningful scores, post hoc test was employed. For the correlation analyses of different parameters, Pearson correlation analysis was used.
RESULTS
UT-II, ET1, and TGFβ1 Levels
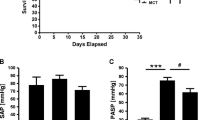
We examined the ET1, UII, and TGF-β1 levels in all groups. The ET1 levels in Groups 1–3 were 5.24 ± 0.40, 6.06 ± 0.39, and 5.70 ± 0.33 pg/ml, respectively. The ET1 levels of Group 2 were significantly higher than those of Group 1 (p < 0.001), and it was significantly diminished in the treatment group (p < 0.04). On the other hand, the UII levels were 3.37 ± 0.19, 4.05 ± 0.14, and 3.70 ± 0.25 ng/ml, respectively, in Groups 1–3, and were significantly diminished in the treatment group (p < 0.001). Finally, the TGF-β1 levels in Groups 1–3 were 58.43 ± 7.93, 73.46 ± 8.28, and 56.70 ± 6.74 ng/ml, respectively, and it was significantly diminished again in the treatment group (p < 0.001) (Fig. 1a–c).

a, b, and c: ET1, UII, and TGF β1 levels in all groups. The asterisks indicates: Group 1 vs. Group 2, p < 0.001; number sign: Group 1 vs. Group 3, p < 0.05; ampersand: Group 2 vs. Group 3, p < 0.05.
Heart Tissue Weight Ratio and Hemodynamic Measures
A tissue weight ratio evaluation of hearts was performed for all groups. RVHI scores were 201.31 ± 35.31 × 10−3, 301.62 ± 39.43 × 10−3, and 246.87 ± 45.19 × 10−3, respectively, for Groups 1–3, and it was significantly decreased in the treatment group (p = 0.001). On the other hand, RVMI scores were 38.16 ± 6.82 × 10−5, 56.10 ± 7.57 × 10−5, and 49.70 ± 5.71 × 10−5, respectively, for Groups 1–3, and that of Group 2 was significantly higher than that of Group 1 (p < 0.001), and there was an insignificant but valuable decrement in the treatment group (p = 0.1). The mPAP scores were 11.10 ± 1.75, 26.87 ± 12.43, and 12.70 ± 2.56, respectively, for Groups 1–3, and those of the treatment group was significantly diminished (p = 0.001) (Fig. 2a–c). The mPAP measurement of a rat from each group is illustrated in Fig. 3.

a, b, and c: RVHI, RVMI, and mPAP. The asterisks indicates Group 1 vs. Group 2, p < 0.001; number sign: Group 1 vs. Group 3, p < 0.05; ampersand: Group 2 vs. Group 3, p < 0.05.

mPAP measurement of a rat from each group.
Histopathology
The microscopic changes were recorded most evidently in Group 2, which were prominent perivascular mononuclear cell infiltration with edema and increment of the connective tissue structures with medial hypertrophy in the vessels of the lung. Interalveolar septum was thickened, and emphysema was detected. It was found that the foregoing lesions in Group 3 ensued with a slight severity. On the other hand, the lungs of Group 1 were histologically normal.
The ocular micrometer measurements in the arteriole and arteries of the lung are summarized in Table 1. When we evaluated the 50–125-μm-diameter arterioles, in contrast to Groups 3 and 1, there was a statistically significant thickness (p < 0.01) in the arteriole walls of Group 2.
On the other hand, when the arterioles’ wall thickness and diameter values were compared, in contrast to Groups 3 and 1, the results were evidently higher in Group 2 (p < 0.001). There was no significant difference between the measurement results of Groups 3 and 1 concerning the arterioles’ wall thickness and diameter (p > 0.05). When we evaluated the arterioles and arteries with diameter greater than 125 μm, the arteriole wall thickness was statistically significant between Groups 1 and 2 (p = 0.01), and there was remarkable, but insignificant decrement, in Group 3 (p > 0.05). The increase in perivascular connective tissue in the arterioles and arteries with diameter greater than 125 μm was mostly observed in Group 2, and the decrease in perivascular connective tissue thickness of arterioles and arteries with diameter greater than 125 μm was statistically significant in Group 3, in contrast to Group 2 (p = 0.001). The histological findings of all groups are presented in Fig. 4.

1A Perivascular connective tissue increment in monocrotaline group, mononuclear cell infiltration, and thickness in interalveolar septum. 1B Slight perivascular mononuclear infiltration and edema in palosuran group. 1C Histological demonstration of lung in control group (H&E, Bar = 100 μm). 2A Severe perivascular edema and mononuclear cell infiltration accompanying medial hypertrophy in the walls of the arterioles between 50- and 125-μm diameters. 2B Slight wall thickness and mononuclear cell infiltration in interalveolar septum and normal demonstration of arterioles between 50- and 125-μm diameters. 2C The arterioles’ wall in the control group (Verhoeff’s Elastic, Bar = 100 μm). 3A Severe perivascular connective tissue increment in the arterioles of higher than 125-μm diameter. 3B Slight perivascular connective tissue increment in the arterioles of higher than 125-μm diameter in palosuran treatment group. 3C Normal image of adventitia layer in the control group (Verhoeff’s Elastic, Bar = 100 μm).
DISCUSSION
In the present study, we tried to demonstrate our hypothesis about UII by antagonizing the peptide with an orally active inhibitor palosuran in an MCT-induced PAH model. With regard to the levels of ET1, UII, and TGF-β1 of Group 2, which was significantly higher than the control (Group 1) and treatment (Group 3) groups, palosuran certainly decreased these peptides’ levels, along with the levels of mPAP, RVHI, and RVMI in the treatment group. We also observed that there was a statistically significant difference between the results of the arteriolar wall thickness and perivascular connective structures of the treatment and patient groups. Our results indicate that palosuran has led to amelioration of the hemodynamic, histological, and biochemical parameters in MCT-induced PAH model, and might be a new therapy alternative for the treatment of PAH.
Although there is no animal model that completely resembles the PAH of humans, the MCT-induced rat model, according to the multiple-hit hypothesis which closely mimics the disease, is the most similar one [19].
The complex pathology of PAH is thought to be based on intimal hyperplasia, medial hypertrophy, adventitial proliferation, thrombosis, and inflammation of the small pulmonary arteries, predominantly with a complicated arteriopathy [20]. Vascular remodeling, including cellular proliferation, differentiation, and delay in apoptosis, with an accelerated extracellular matrix turnover, are on-going processes [21], and the net result is a pan-vasculopathy, where some cytokines, especially TGF-β1 and ET1, could be involved in the pathogenesis. ET1 is a key pathogenic mediator, influencing vasoconstriction, fibrosis, vascular hypertrophy, and inflammation, and is strongly correlated to severity of PAH and prognosis [22, 23].
UII, known as a very potent vasoregulatory peptide, has been shown to be a powerful vasoconstrictor, affecting signaling cascades and gene expression in the cardiovascular and pulmonary systems [24]. It has been proved that pulmonary arterial endothelial and smooth muscles express UII in different models of PAH [25]. As a compelling endogenous peptide within the cardiovascular system, UII might contribute to cardiomyocyte hypertrophy, extracellular matrix production, enhanced vasoconstriction, vascular smooth muscle cell hyperplasia, and endothelial cell (EC) hyper-permeability. Interestingly, UII has been shown to be structurally and functionally mimicking ET1, a vasoconstrictor peptide that plays an important role in the pathophysiology of PAH [26]. However, UII is not a mimicker, but could perhaps be one of the key factors in PAH. This peptide exhibits a pro-angiogenic effect on ECs, resulting in the stimulation of a potent EC activation marker, vascular endothelial growth factor (VEGF). Interestingly, the net result of UII on VEGF was not observed in physiological concentration, but in levels that were demonstrated in vascular pathologies, such as hypertension and atherosclerosis. Additionally, UII directly stimulates fibroblast growth factor, while palosuran inhibits these activation markers by lowering UII. The vasoactive effects of UII on vasculature seem to be in a time-dependent pattern, and in the acute phase of stimulation, the behavior of ECs was found to be directly stimulated. However, a permanent induction with UII resulted in an indirect stimulation of different powerful pro-angiogenics, such as VEGF, ET1, and adrenomedullin [27].
Rho-kinase/ROK/ROCK was identified as one of the effector of the small GTP-binding protein Rho, which plays an important role in various cellular functions. Recently, direct evidence of Rho-kinase activation has been demonstrated in patients with PAH, where its activity was observed to be enhanced in circulating neutrophils and pulmonary arteries of patients with PAH [28]. The animal model-based studies demonstrated that long-term inhibition of Rho-kinase ameliorates MCT-induced PAH. Irrespective of the different etiologies, long-term treatment with Rho-kinase inhibitors ameliorated endothelial dysfunction and suppressed hypercontraction and proliferation of vascular smooth muscle cells and migration of inflammatory cells [29, 30]. A Rho-kinase inhibitor, fasudil, was noted to acutely improve pulmonary hemodynamic in patients with PAH [31–33], and ET1 and UII were observed to lead to similar effects on the pathophysiology of PAH through the Rho-kinase pathway. Most of the previous studies have focused on the vasoconstrictive or vascular smooth muscle proliferative action of UII, and it is argued that this action is mediated by various intracellular signal transduction mechanisms, including phospholipase C, protein kinase C, tyrosine kinases, and the Rho/Rho-kinase related pathways [34–36]. Our results show that ET1 could be a cytokine that is on the downstream of the UII-induced cascade in PAH. The net effect of palosuran by lowering these two angiogenic peptides might indicate that by inhibiting UII, EC-derived on-going stimulus and fibrotic process in the pulmonary arterial and myocardial system could be retrieved. However, preventing angiogenic provocation at any point could reverse all the system to a stabile mode, which could also be an alternative.
According to Mei et al., urantide, a parenteral UII inhibitor, markedly reduced the mPAP levels of MCT-induced PAH in both early and late treatment groups. However, it did not change the MBP. Additionally, Urantide inhibited the pulmonary vascular remodeling remarkably, and the authors concluded that the effect of the drug is through relaxation of the pulmonary arteries [12]. Similarly, in our study, it was demonstrated that palosuran, as a different UII antagonist, significantly decreased the ET1, UII, and TGF-β1 levels, along with mPAP, RVHI, and RVMI in the treatment group. On the other hand, the MBP of all groups were not changed significantly. Additionally, we demonstrated that there was a statistically significant difference between the results of arteriole wall thickness and perivascular connective tissue thickness in the arteries of treatment and patient groups. This difference in the healing effect of palosuran between smaller and larger diameter vessels was explained by the time interval that was not sufficient for adequate treatment.
Nevertheless, there are some limitations of our study. First, we had administered the maximum dose of palosuran to the rats (300 mg/kg), and different dose regimens were ignored. Thus, to determine the optimum dose, further studies are needed. Second, due to budgetary concerns, we underestimated the immunohistochemistry analysis.
In conclusion, owing to its alleviative effect on hemodynamic, histological, and biochemical parameters in MCT-induced PAH, palosuran, as a non-peptidic, orally active, potent, selective, and competitive antagonist of the human UII receptor, may be an optional therapy for the treatment of PAH. However, to verify these findings, determining the optimum dose and addressing other further concerns such as safety and treatment period, new animal models, and clinical experimental studies are required.
References
McLaughlin, V.V., and M.D. McGoon. 2006. Pulmonary arterial hypertension. Circulation 114: 1417–1431.
Kim, N.H.S., and L.J. Rubin. 2002. Endothelin in health and disease: Endothelin receptor antagonists in the management of pulmonary artery hypertension. Journal of Cardiovascular Pharmacology Therapeutics 7: 9–19.
Maguire, J.J., R.E. Kuc, and A.P. Davenport. 2000. Orphan-receptor ligand human urotensin II: Receptor localization in human tissues and comparison of vasoconstrictor responses with endothelin-1. British Journal of Pharmacology 131: 441–446.
Ames, R.S., H.M. Sarau, J.K. Chambers, R.N. Willette, N.V. Aiyar, A.M. Romanic, et al. 1999. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature 401: 282–286.
Hillier, C., C. Berry, M.C. Petrie, P.J. O'Dwyer, C. Hamilton, A. Brown, et al. 2001. Effects of urotensin II in human arteries and veins of varying caliber. Circulation 103: 1378–1381.
Giebing, G., M. Tölle, J. Jürgensen, J. Eichhorst, J. Furkert, M. Beyermann, et al. 2005. Arrestin-independent internalization and recycling of the urotensin receptor contribute to long-lasting urotensin II-mediated vasoconstriction. Circulation Research 97: 707–715.
Djordjevic, T. 2007. Görlach A. Urotensin-II in the lung: A matter for vascular remodelling and pulmonary hypertension? Thrombosis and Haemostasis 98: 952–962.
Maguire, J.J., and A.P. Davenport. 2002. Is urotensin-II the new endothelin? British Journal of Pharmacology 137: 579–588.
Pehlivan, Y., A.M. Onat, G. Comez, and T. Babacan. 2011. Urotensin-II in systemic sclerosis: A new peptide in pathogenesis. Clinical Rheumatology 30: 837–842.
Pehlivan, Y., B. Gogebakan, S. Oztuzcu, M. Ozgen, G.Y. Cetin, R. Bayraktar, et al. 2012. Association between Thr21Met and Ser89Asn polymorphisms of the urotensin II Gene and systemic sclerosis. The Journal of Rheumatology 39: 106–111.
Onat, A.M., I. Turkbeyler, Y. Pehlivan, T. Demir, D.S. Kaplan, S. Taysi, et al. 2010. Urotensin II antogonism with palosuran: A new treatment in pulmonary arterial hypertension. Annals of the Rheumatic Diseases 69(suppl3): 410.
Mei, Y., H. Jin, W. Tian, H. Wang, H. Wang, Y. Zhao, et al. 2011. Urantide alleviates monocrotaline induced pulmonary arterial hypertension in Wistar rats. Pulmonary Pharmacology & Therapeutics 24: 386–393.
Clozel, M., C. Binkert, M. Birker-Robaczewska, C. Boukhadra, S.S. Ding, W. Fischli, et al. 2004. Pharmacology of the urotensin-II receptor antagonist palosuran (ACT-058362; 1-[2-(4-benzyl-4-hydroxy-piperidin-1-yl)-ethyl]-3-(2-methyl- quinolin-4-yl)-urea sulfate salt): First demonstration of a pathophysiological role of the urotensin system. The Journal of Pharmacology and Experimental Therapeutics 311: 204–212.
Alexandru, B., and M.A. Bogdan. 2001. Monocrotaline induces pulmonary hypertension in animal models. Pneumologia 50: 85–89.
Clozel, M., P. Hess, C. Qiu, S.S. Ding, and M. Rey. 2006. The urotensin-II receptor antagonist palosuran improves pancreatic and renal function in diabetic rats. The Journal of Pharmacology and Experimental Therapeutics 316: 1115–1121.
Stinger, R.B., V.J. Iacopino, I. Alter, T.M. Fitzpatrick, J.C. Rose, and P.A. Kot. 1981. Catheterization of the pulmonary artery in the closed-chest rat. Journal of Applied Physiology 51: 1047–1050.
Zhu, X.D., D.M. Shi, Z.C. Jing, Z.C. Jing, L. Li, D. Ma, et al. 2010. The effects of atorvastatin on pulmonary arterial hypertension and expression of p38, p27, and Jab1 in rats. International Journal of Molecular Medicine 26: 541–547.
Varagic, J., E.D. Frohlich, J. Díez, D. Susic, J. Ahn, A. González, et al. 2006. Myocardial fibrosis, impaired coronary hemodynamics, and biventricular dysfunction in salt-loaded SHR. American Journal of Physiology. Heart and Circulatory Physiology 290: 1503–1509.
Robbins, I.M. 2004. Advancing therapy for pulmonary arterial hypertension: Can animal models help? American Journal of Respiratory and Critical Care Medicine 169: 5–6.
McLaughlin, V.V., S.L. Archer, D.B. Badesch, R.J. Barst, H.W. Farber, J.R. Lindner, et al. 2009. American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association: ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the AmericanCollege of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. Journal of the American College of Cardiology 53: 1573–1619.
Essop, M.R. 2010. Contemporary insights into the pathogenesis, diagnosis and therapy of pulmonary arterial hypertension. Cardiovascular Journal of Africa 21: 334–337.
Stewart, D.J., R.D. Levy, P. Cernacek, and D. Langleben. 1991. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Annals of Internal Medicine 114: 464–469.
Rubens, C., R. Ewert, M. Halank, R. Wensel, H.D. Orzechowski, H.P. Schultheiss, et al. 2001. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest 120: 1562–1569.
Djordjevic, T., R.S. BelAiba, S. Bonello, J. Pfeilschifter, J. Hess, and A. Görlach. 2005. Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology 25: 519–525.
Qi, J., J. Du, X. Tang, J. Li, B. Wei, and C. Tang. 2004. The upregulation of endothelial nitric oxide synthase and urotensin-II is associated with pulmonary hypertension and vascular diseases in rats produced by aortocaval shunting. Heart and Vessels 19: 81–88.
MacLean, M.R., D. Alexander, A. Stirrat, M. Gallagher, S.A. Douglas, E.H. Ohlstein, et al. 2000. Contractile responses to human urotensin-II in rat and human pulmonary arteries: Effect of endothelial factors and chronic hypoxia in the rat. British Journal of Pharmacology 130: 201–204.
Albertin, G., D. Guidolin, E. Sorato, R. Spinazzi, A. Mascarin, B. Oselladore, et al. 2009. Pro-angiogenic activity of Urotensin-II on different human vascular endothelial cell populations. Regulatory Peptides 157: 64–71.
Loirand, G., P. Guerin, and P. Pacaud. 2006. Rho kinases in cardiovascular physiology and pathophysiology. Circulation Research 98: 322–334.
Abe, K., H. Shimokawa, K. Morikawa, T. Uwatoku, K. Oi, Y. Matsumoto, et al. 2004. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circulation Research 94: 385–393.
Oka, M., K.A. Fagan, P.L. Jones, and I.F. McMurtry. 2008. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. British Journal of Pharmacology 155: 444–454.
Fukumoto, Y., T. Matoba, A. Ito, H. Tanaka, T. Kishi, S. Hayashidani, et al. 2005. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391–392.
Fujita, H., Y. Fukumoto, K. Saji, K. Sugimura, J. Demachi, J. Nawata, et al. 2010. Acute vasodilator effects of inhaled fasudil, a specific Rhokinase inhibitor, in patients with pulmonary arterial hypertension. Heart and Vessels 25: 144–149.
Ishikura, K., N. Yamada, M. Ito, S. Ota, M. Nakamura, N. Isaka, et al. 2006. Beneficial acute effects of Rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circulation Journal 70: 174–178.
Sauzeau, V., E.L. Mellionnec, J. Bertoglio, E. Scalbert, P. Pacaud, and G. Loirand. 2001. Human urotensin II-induced contraction and arterial smooth muscle cell proliferation are mediated by Rho A and Rho-kinase. Circulation Research 88: 1102–1104.
Watanabe, T., R. Pakala, T. Katagiri, and C.R. Benedict. 2001. Synergistic effect of urotensin II with serotonin on vascular smooth muscle cell proliferation. Journal of Hypertension 19: 2191–2196.
Rossowski, W.J., B.L. Cheng, J.E. Taylor, R. Datta, and D.H. Coy. 2002. Human urotensin II-induced aorta ring contractions are mediated by protein kinase C, tyrosine kinases and Rho-kinase: Inhibition by somatostatin receptor antagonists. European Journal of Pharmacology 438: 159–170.
Acknowledgments
We are very grateful to Actelion Pharmaceuticals Ltd. (Allschwil, Switzerland) for providing palosuran samples.
Conflict of Interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Onat, A.M., Pehlivan, Y., Turkbeyler, I.H. et al. Urotensin Inhibition with Palosuran Could Be a Promising Alternative in Pulmonary Arterial Hypertension. Inflammation 36, 405–412 (2013). https://doi.org/10.1007/s10753-012-9559-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-012-9559-x